

In this issue of Blood, identified thioredoxin-related transmembrane protein 1 (TMX1) as the first antithrombotic vascular thiol isomerase.1
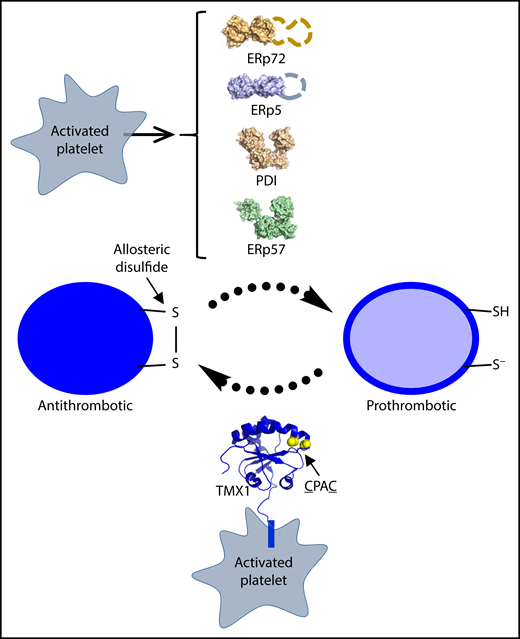
TMX1 is an intrinsic membrane oxidoreductase that is expressed on the activated platelet surface where it negatively regulates thrombosis by oxidizing dithiols to disulfides in αIIbβ3 integrin and likely other proteins.
TMX1 is an intrinsic membrane oxidoreductase that is expressed on the activated platelet surface where it negatively regulates thrombosis by oxidizing dithiols to disulfides in αIIbβ3 integrin and likely other proteins.
Thrombosis is regulated by a cascade of peptide bond cleavages in the clotting factors and vascular cell receptors. Over the last decade, it has become clear that this biology is also controlled by cleavage of the next most frequent covalent bond linking the polypeptide chain, the disulfide bond.2 Disulfide bonds link pairs of cysteine residues in the protein backbone. Although most disulfide bonds serve a structural role in proteins, a subset known as the allosteric disulfides are cleaved in the mature protein to control function.3 The factors that facilitate cleavage (or formation) of the allosteric disulfides involved in thrombosis and hemostasis are the vascular thiol isomerases.4 Protein disulfide isomerase (PDI) and the PDI family members ERp5, ERp57, and ERp72 are secreted by activated platelets and endothelial cells, which is essential for normal thrombosis in mice. Whereas PDI, ERp5, ERp57, and ERp72 are soluble proteins, TMX1 is an intrinsic membrane protein of the platelet plasma membrane (see figure).
TMX1 was originally identified as an endoplasmic reticulum oxidoreductase in a human adenocarcinoma cell line.5 It is a 280–amino acid residue protein consisting of an N-terminal thioredoxin-like domain with an active-site CPAC motif, followed by a transmembrane segment and an intracellular region (see figure). Zhao et al found that platelet activation results in increased levels of TMX1 on the platelet surface, implying that the oxidoreductase can transition from cytoplasmic membranes to the plasma membrane. An important fundamental question is how secretion of TMX1, as well as the other vascular thiol isomerases, is achieved and regulated in platelets. The indications are that a non-classical secretion mechanism is in play. In vitro and in vivo assays of platelet function indicate that TMX1 negatively regulates platelet activity. Knockout of TMX1 in platelets increased platelet incorporation in growing thrombi and shortened tail-bleeding times. This finding contrasts with knockout of the soluble vascular thiol isomerases, which all result in impaired platelet incorporation in thrombi. TMX1, therefore, is the first vascular thiol isomerase with antithrombotic function.
A recurring theme of the vascular thiol isomerases, including TMX1, is their influence on the function of platelet αIIbβ3 integrin. Binding of αIIbβ3 integrin on activated platelets to circulating fibrinogen mediates platelet clumping at sites of vessel injury, and the integrin is an antithrombotic target in acute coronary syndrome. The 4 soluble vascular thiol isomerases bind to β3 integrin. How ERp5 influences αIIbβ3 integrin has been elucidated, but it is still unclear how PDI, ERp57, and ERp72 change integrin function. Platelet ERp5 mediates release of fibrinogen from activated αIIbβ3 integrin by cleaving the β3 subunit C177-C184 disulfide bond.6 Cleavage of this disulfide leads to changes in the ligand binding site that triggers release of fibrinogen. It has been proposed that this chemical event may limit thrombus growth and thus prevent vessel occlusion. TMX1 oxidizes cysteine thiols, presumably to disulfides, in the β3 subunit of αIIbβ3 (see figure). The molecular details of this oxidation and how this influences integrin function are important questions for future studies. It is likely that TMX1 has other, perhaps more important, substrates in the circulation. A kinetic substrate trapping approach, such as that used for PDI in platelet-rich plasma,7 could reveal the TMX1 substrates.
There are now 5 members of the vascular thiol isomerase club that are all required for normal murine thrombosis. One wonders if there are other members and if they work independently or together to control thrombosis. In any case, there is clearly another level of control of thrombosis that we are just starting to uncover. It is possible that the vascular thiol isomerases shuffle electrons among themselves and substrate disulfides.8 They may function as a redox chain in a fashion analogous to that of the Dsb factors in the bacterial periplasm. On the basis of the redox potentials of the active-site dithiols and disulfides, it is conceivable that electrons flow from ERp72 to ERp5 to PDI to ERp57 and to substrate disulfides at any point in the chain. It will be of interest to learn how TMX1 might fit into such a chain. Because TMX1 oxidizes cysteine thiols in αIIbβ3, it is expected to enter nearby ERp57, which is the most oxidizing of the soluble vascular thiol isomerases.
Although our understanding of how the vascular thiol isomerases regulate thrombosis is relatively immature, a plasma membrane–impermeable inhibitor of one of the soluble factors, PDI, is currently being tested as an antithrombotic in a cancer thrombosis clinical trial.9 This is the first of what will likely be many attempts to target this posttranslational modification for the treatment of pathological thrombosis.
Conflict-of-interest disclosure: The author declares no competing financial interests.

