

Key Points
Extracellular TMX1 negatively regulates activation of the αIIbβ3 integrin and platelet aggregation.

Abstract
Secreted platelet protein disulfide isomerases, PDI, ERp57, ERp5, and ERp72, have important roles as positive regulators of platelet function and thrombosis. Thioredoxin-related transmembrane protein 1 (TMX1) was the first described transmembrane member of the protein disulfide isomerase family of enzymes. Using a specific antibody, the recombinant extracellular domain of TMX1 (rTMX1) protein, a knockout mouse model, and a thiol-labeling approach, we examined the role of TMX1 in platelet function and thrombosis. Expression of TMX1 on the platelet surface increased with thrombin stimulation. The anti-TMX1 antibody increased platelet aggregation induced by convulxin and thrombin, as well as potentiated platelet ATP release. In contrast, rTMX1 inhibited platelet aggregation and ATP release. TMX1-deficient platelets had increased aggregation, ATP release, αIIbβ3 activation, and P-selectin expression, which were reversed by addition of rTMX1. TMX1-knockout mice had increased incorporation of platelets into a growing thrombus in an FeCl3-induced mesenteric arterial injury model, as well as shortened tail-bleeding times. rTMX1 oxidized thiols in the αIIbβ3 integrin and TMX1-deficient platelets had increased thiols in the β3 subunit of αIIbβ3, consistent with oxidase activity of rTMX1 against αIIbβ3. Thus, TMX1 is the first identified extracellular inhibitor of platelet function and the first disulfide isomerase that negatively regulates platelet function.
Introduction
Platelets become rapidly activated at the site of vascular injury and have a central role in thrombosis. Of equal importance to pathways that cause platelet activation are those systems that negatively regulate platelets to prevent excessive activation and unwanted thrombosis.1 Platelets have a number of endogenous inhibitors that act at the levels of agonist receptor stimulation, intracellular Ca2+ elevation, and RAP1 activation.1 These cytosolic inhibitors serve to control platelet activation upstream of activation of the αIIbβ3 receptor for fibrinogen and other adhesive proteins.2 Extracellular negative regulators of αIIbβ3 activation have not been well studied.
We and other investigators have shown that several members of the protein disulfide isomerase (PDI) family of enzymes support platelet function and thrombosis via their CGHC active-site motif. These include the prototypical PDI, ERp57, ERp5, and ERp72.3-14 Each of these enzymes is individually required for activation of the αIIbβ3 integrin and platelet aggregation.13 There are no known PDIs that negatively regulate platelet function.
Thioredoxin-related transmembrane protein 1 (TMX1) is a transmembrane member of the PDI family that forms disulfide bonds in newly formed proteins in the endoplasmic reticulum.15,16 These reactions are mediated through a single unique CPAC-active site.15,16 TMX1 preferentially acts on transmembrane polypeptides, including the β1 integrin, while ignoring the same Cys-containing ectodomains if not anchored at the endoplasmic reticulum membrane.16 In the current study, we found that extracellular platelet TMX1 has an unexpected negative regulatory function in platelet activation and thrombosis.
Study design
Generation and characterization of TMX1-deficient mice and the recombinant extracellular domain of TMX1 (rTMX1) protein are described in the supplemental Materials and methods (available on the Blood Web site). RNA extraction, reverse-transcription polymerase chain reaction (RT-PCR), polymerase chain reaction, western blotting, coagulation assays, bleeding times, flow cytometry, platelet aggregation/secretion, FeCl3-induced thrombosis, PDI assays, labeling of platelet αIIbβ3 with 3-(N-maleimidylpropionyl)-biocytin, and statistical analysis were described previously13 and are included in the supplemental Materials and methods.
Labeling of thiols in αIIbβ3 with iodoTMT
αIIbβ3 (1.5 μg; R&D Systems) was treated with TCEP Disulfide Reducing Resin for 60 minutes at room temperature and then incubated with 5 μM rTMX1 for 20 minutes at 37°C. IodoTMT (400 μM) was added, and thiols were labeled in phosphate buffered saline containing 1% sodium dodecyl sulfate and 5 mM EDTA at pH 7.4 for 1 hour at 37°C. Blotting was performed using anti-TMT, rabbit anti-β3, monoclonal anti-αIIb, and monoclonal anti-TMX1 antibodies.
Results and discussion
Activated platelets express TMX1 on the membrane surface
Human platelets express TMX1 messenger RNA (mRNA), as detected by quantitative RT-PCR (supplemental Figure 1A). On blotting, a polyclonal anti-TMX1 antibody recognized a single band the size of the TMX1 protein (32 kDa) in human platelets and endothelial cells, whereas leukocytes contain little TMX1 (supplemental Figure 1B). The antibody does not react with other CXYC-containing transmembrane PDIs, TMX3 and TMX4 (supplemental Figure 1C). Using this antibody, TMX1 was found to increase on the platelet surface with thrombin-induced activation (Figure 1A), suggesting that TMX1 is externalized from the platelet-dense tubular system as other PDIs.17
![Figure 1. Extracellular platelet TMX1 inhibits aggregation of human platelets. (A) TMX1 expression on the platelet surface increases with platelet activation. TMX1 expression on nonactivated (NA) and thrombin-activated platelets. Representative line graph (left panel) and combined results (right panel). Data are mean ± standard error of the mean (SEM), n = 5 for each group. Human platelets were incubated with 1 U/mL thrombin for 5 minutes, followed by flow cytometry measurement using the anti-TMX1 antibody B01P. The anti-TMX1 antibody B01P increases platelet aggregation induced by SFLLRN (B), convulxin (C), and thrombin (D). In these studies, human platelets were incubated with IV.3 (10 μg/mL) for 5 minutes and then with B01P (20 μg/mL) for 5 minutes, after which they were activated. Representative tracings for aggregation and ATP release (left panels) and the combined results (right panels) are shown; data are mean ± SEM, n = 3 (B) and n = 4 (C-D). rTMX1 containing the CPAC-motif (rTMX1) inhibits convulxin-induced platelet aggregation (E) and thrombin-induced platelet aggregation and ATP release (F). Human platelets were incubated with rTMX1 for 5 minutes at the indicated concentrations before stimulation. Representative aggregation and ATP release (left panels) and combined results (right panels); data are mean ± SEM, n = 4 for each group. (G) Inactivated rTMX1 [TMX1(oo)] increases convulxin-induced platelet aggregation. Representative aggregation (left panel) and combined results (right panel); data are mean ± SEM, n = 6 for each group. *P < .05, **P < .01, ***P < .001, Student t test. IgG, normal mouse immunoglobulin G; MFI, mean fluorescence intensity.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/3/10.1182_blood-2018-04-844480/7/m_blood844480f1.png?Expires=1769195912&Signature=IQNf7TSq8AphkDakFGNkUJgAuNyxMcj3aWxOd3IqSg2R3um9qChPIAX0Dweet7ulUsPVCzD9rWWBjr55heLfqGIzQ84g8kjF73kpxiLjSX1s7k33UNQPMTchsYmP7-erJdJju0jSvunlOrhJyxJKVnVVov3EDNjPq7lUnwIv4F8nNTCgCozi7ZidpcJb9RAOvX80QrItRZJaH4MNzFra9iQhVLU1L0fiyW-qayLesnZw~ud014wh8VWxBKhIsDBSTYRdY4m-dHrujMR6AvQrzFBNb7S1mHdkyp6PX8CRUaJ21niJzzoN0kAy7TBiAJZfyzI0RobD-eWE0uppEHAuAw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Extracellular platelet TMX1 inhibits aggregation of human platelets. (A) TMX1 expression on the platelet surface increases with platelet activation. TMX1 expression on nonactivated (NA) and thrombin-activated platelets. Representative line graph (left panel) and combined results (right panel). Data are mean ± standard error of the mean (SEM), n = 5 for each group. Human platelets were incubated with 1 U/mL thrombin for 5 minutes, followed by flow cytometry measurement using the anti-TMX1 antibody B01P. The anti-TMX1 antibody B01P increases platelet aggregation induced by SFLLRN (B), convulxin (C), and thrombin (D). In these studies, human platelets were incubated with IV.3 (10 μg/mL) for 5 minutes and then with B01P (20 μg/mL) for 5 minutes, after which they were activated. Representative tracings for aggregation and ATP release (left panels) and the combined results (right panels) are shown; data are mean ± SEM, n = 3 (B) and n = 4 (C-D). rTMX1 containing the CPAC-motif (rTMX1) inhibits convulxin-induced platelet aggregation (E) and thrombin-induced platelet aggregation and ATP release (F). Human platelets were incubated with rTMX1 for 5 minutes at the indicated concentrations before stimulation. Representative aggregation and ATP release (left panels) and combined results (right panels); data are mean ± SEM, n = 4 for each group. (G) Inactivated rTMX1 [TMX1(oo)] increases convulxin-induced platelet aggregation. Representative aggregation (left panel) and combined results (right panel); data are mean ± SEM, n = 6 for each group. *P < .05, **P < .01, ***P < .001, Student t test. IgG, normal mouse immunoglobulin G; MFI, mean fluorescence intensity.
Extracellular platelet TMX1 inhibits aggregation of human platelets. (A) TMX1 expression on the platelet surface increases with platelet activation. TMX1 expression on nonactivated (NA) and thrombin-activated platelets. Representative line graph (left panel) and combined results (right panel). Data are mean ± standard error of the mean (SEM), n = 5 for each group. Human platelets were incubated with 1 U/mL thrombin for 5 minutes, followed by flow cytometry measurement using the anti-TMX1 antibody B01P. The anti-TMX1 antibody B01P increases platelet aggregation induced by SFLLRN (B), convulxin (C), and thrombin (D). In these studies, human platelets were incubated with IV.3 (10 μg/mL) for 5 minutes and then with B01P (20 μg/mL) for 5 minutes, after which they were activated. Representative tracings for aggregation and ATP release (left panels) and the combined results (right panels) are shown; data are mean ± SEM, n = 3 (B) and n = 4 (C-D). rTMX1 containing the CPAC-motif (rTMX1) inhibits convulxin-induced platelet aggregation (E) and thrombin-induced platelet aggregation and ATP release (F). Human platelets were incubated with rTMX1 for 5 minutes at the indicated concentrations before stimulation. Representative aggregation and ATP release (left panels) and combined results (right panels); data are mean ± SEM, n = 4 for each group. (G) Inactivated rTMX1 [TMX1(oo)] increases convulxin-induced platelet aggregation. Representative aggregation (left panel) and combined results (right panel); data are mean ± SEM, n = 6 for each group. *P < .05, **P < .01, ***P < .001, Student t test. IgG, normal mouse immunoglobulin G; MFI, mean fluorescence intensity.
TMX1 is a negative regulator of platelet aggregation
Preincubation of platelets with the anti-TMX1 antibody increased platelet aggregation induced by SFLLRN, convulxin, and thrombin (Figure 1B-D) and increased ATP release (Figure 1D). The antibody inhibited the oxidase activity of rTMX115 but did not itself induce aggregation or enhance aggregation of TMX1-null platelets (characterized in “Generation and characterization of TMX1-deficient mice”), confirming specificity for TMX1 on platelets (supplemental Figure 1D-F). rTMX1 inhibited convulxin and thrombin-induced platelet aggregation (Figure 1E-F), as well as thrombin-induced ATP release (Figure 1E), whereas inactivated rTMX1 potentiated convulxin-induced aggregation (Figure 1G). In contrast, rTMX3 (the recombinant extracellular form of another transmembrane PDI found in platelets),18 did not inhibit aggregation, whereas inactivated rTMX3 did (supplemental Figure 2). These data suggest that TMX1 is a negative regulator of platelet aggregation mediated by GPVI and thrombin receptors.
Additional studies showed that rTMX1 inhibited the binding of the monovalent fibrinogen γ-chain to convulxin-activated platelets (supplemental Figure 3), implying that TMX1 inhibits a conformational change in αIIbβ3. Although rTMX1 inhibited aggregation (Figure 1E-F), it did not affect the association of αIIbβ3 with talin-1 and kindlin-3 (supplemental Figure 4). rTMX1 did not affect thrombin-induced ATP release of β3-null platelets (supplemental Figure 5), suggesting that the negative regulation of ATP release by TMX1 depends on αIIbβ3.
Generation and characterization of TMX1-deficient mice
To further study the role of TMX1 in platelet function, TMX1-knockout mice were generated, using the knockout-first conditional-ready strategy.19 The germline-transmitted targeted allele was confirmed by genotyping of TMX1-deficient (TMX1−/−, knockout-first) mice (supplemental Figure 6A). RT-PCR confirmed the absence of TMX1 mRNA in platelets (supplemental Figure 6B). The mRNA levels of PDI, ERp57, and ERp72 were comparable to those of wild-type mice, indicating successful targeting of TMX1. Platelet counts were comparable to those of wild-type TMX1+/+ control mice (supplemental Figure 6C). Resting platelets from TMX1−/− mice had normal expression of the major platelet surface glycoproteins (αIIbβ3, GPVI, and GPIb) and P-selectin and did not have activation of αIIbβ3 (supplemental Figure 6D). Prothrombin time and partial thromboplastin time were normal in TMX1−/− mice (supplemental Figure 6E-F), consistent with these mice not having a coagulopathy.
TMX1 deficiency potentiates platelet function, thrombosis, and hemostasis in mice
Convulxin, thrombin, and collagen-induced aggregation, ATP release, activation of αIIbβ3, and P-selectin expression all increased in TMX1-deficient platelets (Figure 2A-E; supplemental Figure 7). rTMX1 reversed these processes (Figure 2C-E). Incorporation of platelets into the thrombus in TMX1−/− mice was substantially increased in an FeCl3-induced mesenteric artery injury model (Figure 2F), with the increase being statistically significant over the 9- to 15-minute time interval (Figure 2G). TMX1−/− mice also had significantly shorter tail bleeding times than TMX1+/+ littermate control mice (Figure 2H).
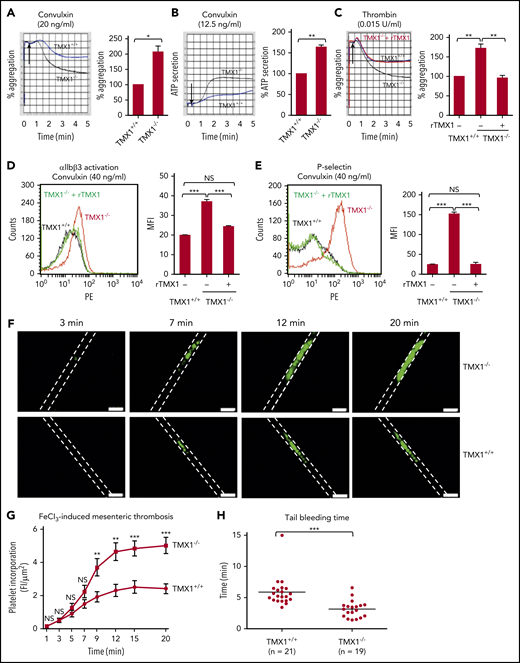
Deficiency in TMX1 potentiates platelet function and thrombosis and shortens bleeding times in mice. Convulxin-induced (A) and thrombin-induced (C) aggregation and convulxin-induced ATP release (B) for platelets from wild-type (TMX1+/+) and TMX1-deficient (TMX1−/−) mice. (C) rTMX1 (2 μM) was added to TMX1-null platelets 5 minutes prior to platelet activation. Representative tracings (left panels) and combined results (right panel); data are mean ± standard error of the mean (SEM), n = 4 (A-B), n = 5 (C). *P < .05, **P < .01, Student t test (A-B), analysis of variance (C). TMX1-deficient platelets have enhanced convulxin-induced activation of αIIbβ3 (detected by the JON/A activation-dependent antibody) (D) and P-selectin expression (E). Adding rTMX1 (2 μM) 5 minutes prior to platelet activation reverses the increased aggregation and P-selectin expression. Representative line graphs (left panels) and combined results (right panels); data are mean ± SEM, n = 4. Platelets from wild-type littermate control or TMX1-deficient mice were stimulated with convulxin for 5 minutes, followed by flow cytometry analysis. ***P < .001, analysis of variance. (F) Incorporation of platelets into a growing thrombus in TMX1+/+ and TMX1−/− mice was detected by Alexa Fluor–488 anti-CD41 after an FeCl3 (3.5%)–induced mesenteric arterial injury. Images were taken at 3, 7, 12, and 20 minutes. Dashed lines mark the vessel wall. Scale bars, 200 μm. Mean vessel diameters: TMX1+/+ mice, 94.34 ± 3.20 μm; TMX1−/− mice, 93.71 ± 5.71 μm (P = not significant). (G) Composite of fluorescent intensity per area (FI/μm2) in TMX1+/+ mice (n = 13 from 4 mice) and TMX1−/− mice (n = 13 from 3 mice); data are mean ± SEM. **P < .01, ***P < .001, Student t test. (H) Tail bleeding time in TMX1+/+ and TMX1−/− mice; data are mean ± SEM. ***P < .001, Student t test. NS, not significant.
Deficiency in TMX1 potentiates platelet function and thrombosis and shortens bleeding times in mice. Convulxin-induced (A) and thrombin-induced (C) aggregation and convulxin-induced ATP release (B) for platelets from wild-type (TMX1+/+) and TMX1-deficient (TMX1−/−) mice. (C) rTMX1 (2 μM) was added to TMX1-null platelets 5 minutes prior to platelet activation. Representative tracings (left panels) and combined results (right panel); data are mean ± standard error of the mean (SEM), n = 4 (A-B), n = 5 (C). *P < .05, **P < .01, Student t test (A-B), analysis of variance (C). TMX1-deficient platelets have enhanced convulxin-induced activation of αIIbβ3 (detected by the JON/A activation-dependent antibody) (D) and P-selectin expression (E). Adding rTMX1 (2 μM) 5 minutes prior to platelet activation reverses the increased aggregation and P-selectin expression. Representative line graphs (left panels) and combined results (right panels); data are mean ± SEM, n = 4. Platelets from wild-type littermate control or TMX1-deficient mice were stimulated with convulxin for 5 minutes, followed by flow cytometry analysis. ***P < .001, analysis of variance. (F) Incorporation of platelets into a growing thrombus in TMX1+/+ and TMX1−/− mice was detected by Alexa Fluor–488 anti-CD41 after an FeCl3 (3.5%)–induced mesenteric arterial injury. Images were taken at 3, 7, 12, and 20 minutes. Dashed lines mark the vessel wall. Scale bars, 200 μm. Mean vessel diameters: TMX1+/+ mice, 94.34 ± 3.20 μm; TMX1−/− mice, 93.71 ± 5.71 μm (P = not significant). (G) Composite of fluorescent intensity per area (FI/μm2) in TMX1+/+ mice (n = 13 from 4 mice) and TMX1−/− mice (n = 13 from 3 mice); data are mean ± SEM. **P < .01, ***P < .001, Student t test. (H) Tail bleeding time in TMX1+/+ and TMX1−/− mice; data are mean ± SEM. ***P < .001, Student t test. NS, not significant.
Previous studies showed a role for thiols in αIIbβ3 in the activation of this integrin and platelet aggregation.20-24 To examine the effect of TMX1 on redox regulation of αIIbβ3, rTMX1 was incubated with native αIIbβ3 integrin or with αIIbβ3 reduced by TCEP, and the effect of adding rTMX1 on thiol labeling was examined. Little labeling of thiols was detected in untreated αIIbβ3 (supplemental Figure 8A, lane 1), although longer exposures detected thiols. Addition of rTMX1 had no effect on thiol labeling of αIIbβ3 (supplemental Figure 8A, lane 2), although rTMX1 itself contained labeled thiols (supplemental Figure 8A, bottom, lane 2). In contrast, both subunits of TCEP-treated αIIbβ3 contained thiols, which were lost with the addition of rTMX1 (supplemental Figure 8A, lanes 3 and 4), suggesting that rTMX1 oxidizes thiols in αIIbβ3. In contrast to the oxidase activity of rTMX1 (supplemental Figure 1D), rTMX1 had limited reductase activity compared with PDI (supplemental Figure 8B-C). Almost 90% of the CPAC active site is in the disulfide form (supplemental Figure 8D), consistent with oxidase activity. TMX1 deficiency in platelets increased thiols in the β3 subunit of αIIbβ3 (supplemental Figure 8E). These data are consistent with TMX1 having an oxidase function in platelets.
These studies provide 3 lines of evidence that extracellular platelet TMX1 negatively regulates platelet function. First, an antibody to TMX1 that recognizes TMX1 on the platelet surface potentiates platelet aggregation (with blockade of the FcγRIIa receptor). This directly contrasts with the effect of antibodies on the other platelet PDIs; anti-PDI,25-27 anti-ERp5,11,12 anti-ERp57,7,8 and anti-ERp7214 all inhibit platelet aggregation. Second, addition of rTMX1 inhibits aggregation, whereas inactivated rTMX1 potentiates aggregation. This directly contrasts with the addition of PDI, ERp57, or ERp72 to human platelets, all of which potentiate platelet aggregation, whereas the inactivated enzymes inhibit aggregation.6,9,13 Third, TMX1 deficiency enhances platelet aggregation. This contrasts with the effect of deletion of PDI, ERp57, or ERp72 in platelets, which decreases aggregation.5,6,9,13 The decrease is recovered by the addition of the missing PDI, whereas addition of rTMX1 reverses the enhanced aggregation of TMX1-null platelets (Figure 2C). Thus, anti-TMX1 and TMX1 deletion potentiate platelet aggregation, whereas addition of rTMX1 inhibits platelet aggregation, implicating TMX1 as a negative regulator of platelet function.
Although endogenous inhibitors of earlier platelet activation events balance the process of platelet activation,1 TMX1 appears to inhibit it at the final common pathway of activation of αIIbβ3. TMX1 may serve to counterbalance PDI, ERp57, ERp5, and ERp72, all of which potentiate activation of αIIbβ3. Although thiols in αIIbβ3 have a role in activation of this receptor,20-23 the negative-regulatory role of TMX1 may occur via oxidizing thiols in this integrin. These findings provide a biological basis for studies of TMX1 in disease states and may provide new strategies to treat bleeding or thrombosis.
Data on the functional activity of rTMX1, binding of the γ chain of fibrinogen to activated platelets, and labeling of thiols in aIIbβ3 can be found in the supplemental Materials and methods.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grant R01 HL118526 (D.W.E.), the National Natural Science Foundation of China (81770138, 91539122, and 81670133), and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Authorship
Contribution: Y.W. designed the project, supervised and performed research, analyzed data, and wrote the manuscript; Z.Z, J.Z., F.C., and A.Y. performed research and collected and analyzed data; and D.W.E. designed the project, supervised the research, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Yi Wu, The Cyrus Tang Hematology Center, Jiangsu Institute of Hematology, First Affiliated Hospital, Soochow University, 199 Ren-Ai Rd, Suzhou 215123, China; e-mail: yiwu99@gmail.com; and David W. Essex, Temple University School of Medicine, Room 204 MRB, 3420 North Broad St, Philadelphia, PA 19140; e-mail: david.essex@temple.edu.


