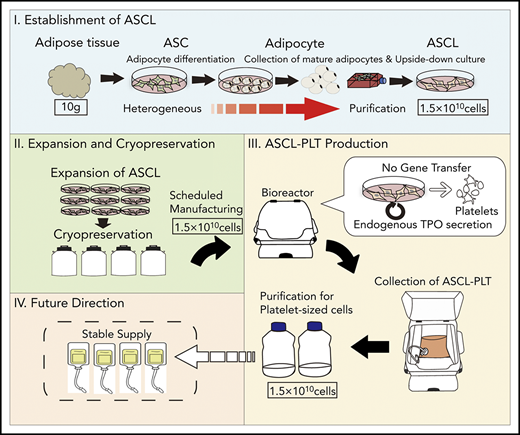
Key Points
Platelets were manufactured on a large scale from a novel human ASCL as a donor-independent source.
ASCL-PLTs have similar characteristics to those of peripheral platelets and might have an additional function as mesenchymal-like cells.

Abstract
The clinical need for platelet transfusions is increasing; however, donor-dependent platelet transfusions are associated with practical problems, such as the limited supply and the risk of infection. Thus, we developed a manufacturing system for platelets from a donor-independent cell source: a human adipose-derived mesenchymal stromal/stem cell line (ASCL). The ASCL was obtained using an upside-down culture flask method and satisfied the minimal criteria for defining mesenchymal stem cells (MSCs) by The International Society for Cellular Therapy. The ASCL showed its proliferation capacity for ≥2 months without any abnormal karyotypes. The ASCL was cultured in megakaryocyte induction media. ASCL-derived megakaryocytes were obtained, with a peak at day 8 of culture, and ASCL-derived platelets (ASCL-PLTs) were obtained, with a peak at day 12 of culture. We observed that CD42b+ cells expressed an MSC marker (CD90) which is related to cell adhesion. Compared with peripheral platelets, ASCL-PLTs exhibit higher levels of PAC1 binding, P-selectin surface exposure, ristocetin-induced platelet aggregation, and ADP-induced platelet aggregation, as well as similar levels of fibrinogen binding and collagen-induced platelet aggregation. ASCL-PLTs have lower epinephrine-induced platelet aggregation. The pattern of in vivo kinetics after infusion into irradiated immunodeficient NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ mice was similar to that of platelet concentrates. ASCL-PLTs have similar characteristics to those of peripheral platelets and might have an additional function as MSCs. The establishment of the ASCL and its differentiation into ASCL-PLTs do not require gene transfer, and endogenous thrombopoietin is used for differentiation. The present protocol is a simple method that does not require feeder cells, further enhancing the clinical application of our approach.
Introduction
Platelets are essential for hemostatic plug formation. Platelet transfusions are widely used for patients with severe thrombocytopenia that is caused by malignancy, chemotherapy, immune disorders, infections, or inherited platelet disorders, as well as for other indications.1-3 The clinical need for platelet transfusions is increasing, with >4.5 million platelet units transfused (>2.4 × 1011 platelets per unit) per year in Europe and the United States.3 However, platelet transfusions are donor dependent and associated with severe problems, such as a limited supply because of their short shelf life (eg, 5 days in the United States) and the risk of bacterial infection and serious immune reactions. Therefore, the development of an efficient production system for platelets from a donor-independent source is crucial.
Platelets are released from megakaryocytes (MKs) through several steps, beginning with stem cells. Stem cells differentiate into immature MKs and further into mature MKs with unique features, such as a large size and polyploidism.4-7 Finally, mature MKs release platelets. The lineage commitment is primarily regulated by the cytokine thrombopoietin (TPO) and transcription factors, such as p45NF-E2.8-13 MKs and subsequent platelets have been produced from hematopoietic stem cells (HSCs), embryonic stem cells, induced pluripotent stem (iPS) cells, endometrial stromal cells, and fibroblasts transfected with a combination of p45NF-E2, Maf G, and Maf K when these cells were cultured in defined media in the presence of recombinant TPO (rTPO).2,8,14-25 Among these cells as a source of nondonor-derived platelets, HSCs are limited by their low availability, and embryonic stem cells and iPS cells require sophisticated experimental techniques to differentiate into MKs and platelets. Additionally, the use of iPS cells and p45NF-E2/Maf–expressing fibroblasts requires gene transfer to induce their differentiation into MK lineages. Platelets contain RNA and synthesize proteins26 ; thus, the mutagenic potential of manufactured platelets must be investigated. However, the appropriate cell sources and culture methods for clinical transfusion remain controversial.
Adipose-derived stromal cells (ASCs), also known as preadipocytes, are an attractive candidate cell source for the manufacture of platelets for clinical application.27-31 First, their differentiation does not require gene transfer, because ASCs contain important genes related to MK differentiation and platelet production, such as p45NF-E2, GATA2, and RUNX1.28 ASCs use endogenous genes to differentiate into MKs and functional platelets. Second, ASCs differentiate into MKs and subsequently produce platelets utilizing endogenous TPO and its receptor, c-MPL.31 ASCs secrete endogenous TPO via interactions between transferrin and its receptor, CD71.31 Because other stem cells, including HSCs and iPS cells, require the addition of rTPO to differentiate into MK lineages, ASCs have an advantage for platelet manufacture; however, ASCs contain heterogeneous cells and are not suitable as a donor-independent cell source. Based on these observations in basic research, we have tried to manufacture clinically available platelets without gene transfer by establishing a human adipose-derived mesenchymal stromal/stem cell line (ASCL). In the present study, the ASCL shows the minimal criteria for defining mesenchymal stem cells (MSCs) by The International Society for Cellular Therapy.32,33 ASCL-derived cells cultured in MK lineage induction (MKLI) media have similar characteristics to those of CD34+ cord blood cell–derived MKs (CD34+CB-MKs) and additional characteristics normally observed in MSC population. Thus, we distinguished between HSC-derived MKs and platelets and ASCL-derived MKs (ASCL-MKs) and ASCL-derived platelets (ASCL-PLTs). The functions of ASCL-PLTs obtained by large-scale culture and in vivo kinetics were also compared between ASCL-PLTs and platelet concentrates from the Japanese Red Cross Society. Agonist-induced platelet aggregation was higher on ASCL-PLTs than on platelet concentrates. The pattern of in vivo kinetics and hemostatic function after infusion into irradiated immunodeficient NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice was similar to that of platelet concentrates.
Methods
Cell culture
Subcutaneous adipose tissue was obtained from patients undergoing reconstruction surgery after tumorectomy at Keio University Hospital (Tokyo, Japan). All patients gave written informed consent, and the study was approved by the Keio University Research Ethics Committee. ASCs were cultured with Adipocyte Differentiation Medium (Cell Applications), and fat drops from trypsinized cells underwent an upside-down culture.34-36
The ASCL was cultured in MKLI media in the absence of rTPO to obtain MKs and platelets.
Microarray analysis
Genes related to MSCs, which show their multipotency and include 58 key genes, were analyzed using microarray.37-41
Additional methods are described in supplemental Materials, available on the Blood Web site.
Results
Characterization of a novel human ASCL
To obtain an ASCL as a donor-independent cell source for the large-scale production of platelets, we modified the established method for dedifferentiating a fat drop into preadipocytes/ASCs using the upside-down culture flask method.34-36 Instead of using a large volume of fat, as described in the original method, we developed the ASCL from a small number of ASCs (Figure 1A), because ASCs are able to expand. We used 0.5 to 1 g of subcutaneous adipose tissue to generate the ASCL. The morphologic change from fat drops into adherent cells indicates that an ASCL was obtained (supplemental Figure 1). We characterized the ASCL based on its proliferation ability, surface markers, ability to differentiate into mesenchymal cells, and karyotype. The ASCL retained proliferative ability for ≥2 months (n = 3) (Figure 1B). The ASCL satisfied the minimal criteria for defining MSCs by The International Society for Cellular Therapy32,33 : (1) adherence to plastic in in vitro culture (supplemental Figure 1A), (2) ability to differentiate into osteoblasts, mature adipocytes, and chondrocytes in vitro (Figure 1C), and (3) expression (>95% of cells) of CD73, CD90, and CD105 and no expression (<2% of cells) of CD45, CD34, CD14, CD11b, CD19, or HLA-DR. We also examined other markers for mesenchymal cells and hematopoietic cells. The ASCL expressed the mesenchymal cell markers CD13, CD29, CD44, CD71, CD166, and HLA-ABC but not the hematopoietic cell markers CD41, CD42b, and CD56 (Figure 1D). We then performed a genetic analysis of the ASCL after 2 months of culture. Karyotype analysis revealed no abnormalities in the ASCL (Figure 1E).
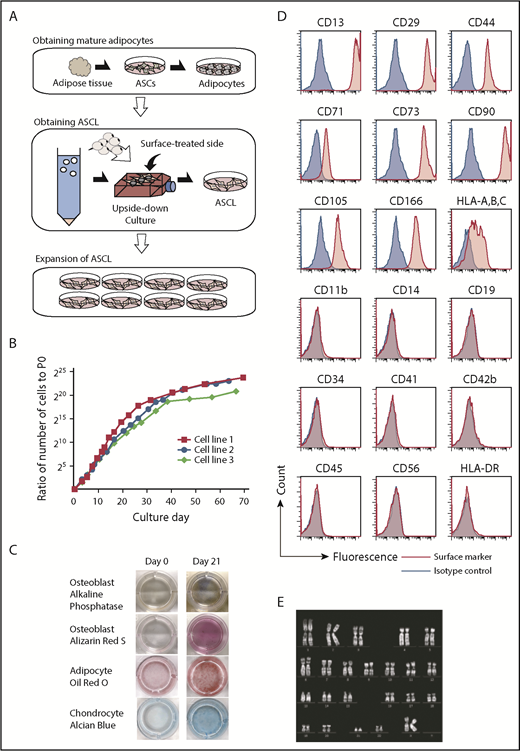
Characterization of a novel human ASCL. (A) Schema shows the process to establish an ASCL from human subcutaneous adipose tissue. Adipose tissue (0.5-1 g) digested with collagenase was centrifuged to obtain ASCs. ASCs were cultured in conditioned media to differentiate into mature adipocytes. Fat drops from trypsinized cells were moved to an upside-down culture. Floating mature adipocytes with fat droplets were collected and placed into the flask, which was completely filled with the media. In the flask completely filled with medium over 14 days without medium change, mature adipocytes with a fat droplet on the flask surface changed into the ASCL. A morphological change from a fat drop into adherent cells indicates that an ASCL is obtained. (B) A proliferation assay was performed on an ASCL established from 3 subjects. (C) The ASCL differentiated into osteoblasts, mature adipocytes, and chondrocytes. The ASCL was cultured in conditioned media for each differentiation, and we assessed their capacity for differentiation using cell staining. (D) Expression of surface markers in the ASCL was estimated by flow cytometry. (E) Representative data for karyotype analysis of the ASCL.
Characterization of a novel human ASCL. (A) Schema shows the process to establish an ASCL from human subcutaneous adipose tissue. Adipose tissue (0.5-1 g) digested with collagenase was centrifuged to obtain ASCs. ASCs were cultured in conditioned media to differentiate into mature adipocytes. Fat drops from trypsinized cells were moved to an upside-down culture. Floating mature adipocytes with fat droplets were collected and placed into the flask, which was completely filled with the media. In the flask completely filled with medium over 14 days without medium change, mature adipocytes with a fat droplet on the flask surface changed into the ASCL. A morphological change from a fat drop into adherent cells indicates that an ASCL is obtained. (B) A proliferation assay was performed on an ASCL established from 3 subjects. (C) The ASCL differentiated into osteoblasts, mature adipocytes, and chondrocytes. The ASCL was cultured in conditioned media for each differentiation, and we assessed their capacity for differentiation using cell staining. (D) Expression of surface markers in the ASCL was estimated by flow cytometry. (E) Representative data for karyotype analysis of the ASCL.
Differentiation of the ASCL into ASCL-MKs
To obtain ASCL-MKs, the ASCL was cultured in MKLI media. ASCL-MKs were characterized by flow cytometric analysis and immunostaining. The frequency of CD41 and CD42b, specific surface markers for MK lineages, was analyzed during culture in MKLI media for 14 days31 (Figure 2A). Data for the isotype control are shown in supplemental Figure 2A. Before MK induction (day 0), the cells did not express these surface markers (Figure 1D). The frequency of CD41-expressing cells increased, with a peak at day 8 (63.2 ± 1.0%, n = 3; Figure 2A), and then decreased slightly. The frequency of CD41/CD42b-expressing cells also peaked at day 8 (15.0 ± 0.3%, n = 3; Figure 2A). The expression of CD42b was not dependent on the culture period. We observed that the ASCL derived from different individuals did not differ with regard to MK yield (supplemental Figure 2B).
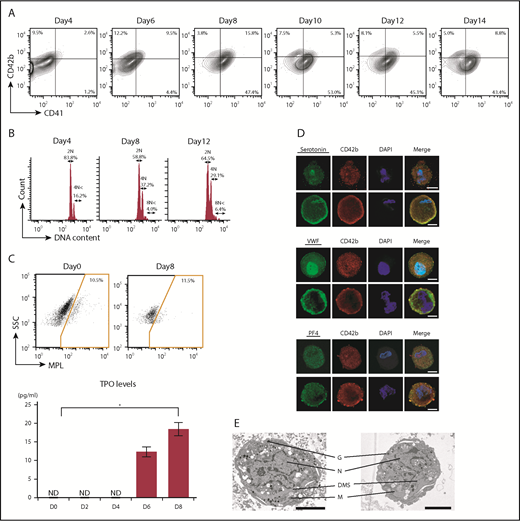
Differentiation of the ASCL into ASCL-MKs. (A) Representative data for CD41/CD42b expression in ASCL-MKs (n = 3). (B) Representative data for DNA ploidy in ASCL-MKs. (C) Representative data for c-MPL surface expression in the ASCL and TPO secretion levels during MK differentiation from the ASCL (n = 2). *P < .01. (D) Expression of serotonin, VWF, and PF4 in MKs derived from the ASCL cultured in MKLI media for 8 days. Cells stained with the antibody for CD42b (red), for DAPI (blue), and for serotonin, VWF, or PF4 (green). Scale bars, 10 μm. (E) Electron microscopic images of ASCL-MKs. Scale bars, 5 μm. DMS, demarcation membrane system; G, granule; M, mitochondrion; N, nucleus.
Differentiation of the ASCL into ASCL-MKs. (A) Representative data for CD41/CD42b expression in ASCL-MKs (n = 3). (B) Representative data for DNA ploidy in ASCL-MKs. (C) Representative data for c-MPL surface expression in the ASCL and TPO secretion levels during MK differentiation from the ASCL (n = 2). *P < .01. (D) Expression of serotonin, VWF, and PF4 in MKs derived from the ASCL cultured in MKLI media for 8 days. Cells stained with the antibody for CD42b (red), for DAPI (blue), and for serotonin, VWF, or PF4 (green). Scale bars, 10 μm. (E) Electron microscopic images of ASCL-MKs. Scale bars, 5 μm. DMS, demarcation membrane system; G, granule; M, mitochondrion; N, nucleus.
DNA ploidy, a hallmark of MKs, ranged from 2N to 4N in CD41+ cells on day 4 (Figure 2B). We observed DNA ploidy >8N in CD41+ cells on days 8 and 12 (Figure 2B). The ASCL derived from different individuals did not differ in DNA ploidy by >8N on day 8 (4.4% for lot 2 and 7.5% for lot 3). Also, the CD41− cells cultured in MK induction media did not have a high-ploidy population. CD41+ cells had a high-ploidy population (supplemental Figure 2C). Similar to ASCL-MKs, CD34+CB-MKs showed DNA polyploidy: 69.8 ± 4.0% for 2N, 28.7 ± 4.0% for 4N, and 1.5 ± 0.3% for >8N (n = 3). Our findings for CD34+CB-MKs were similar to those of Mattia et al.42
Previously, we reported that ASCs express c-MPL. When ASCs were cultured in MKLI media, ASCs secreted endogenous TPO to drive MK lineages.31 Similar to ASCs, the ASCL expressed c-MPL and secreted TPO during MK differentiation (Figure 2C). Data of isotype control is shown in supplemental Figure 2D. Expression of the MK-related intracellular factors serotonin, VWF, and PF4 was also observed in ASCL-MKs by immunostaining (Figure 2D). More images of MK-related intracellular factors in ASCL-MKs and CD34+CB-MKs are shown in supplemental Figure 2E and 2G. We also observed CD49b, a collagen receptor in platelets, in ASCL-MKs (supplemental Figure 2F). We performed an enzyme-linked immunosorbent assay (ELISA) to quantify the presence of serotonin, VWF, and PF4 in 1 × 106 ASCL-MKs and 1 × 106 CD34+CB-MKs. The expression of serotonin is shown in Figure 2D and supplemental Figure 2E-G. The actual value of serotonin in 1 × 106 CD34+CB-MKs was below the limit of detection for the ELISA and was 18 ± 3.8 pg in 1 × 106 ASCL-MKs. The amount of VWF in CD34+CB-MKs was higher than that in ASCL-MKs, and the amount of PF4 was similar between CD34+CB-MKs and ASCL-MKs (supplemental Figure 2H). We also examined the expression and quantification of VEGF, as an α granule, in 1 × 106 CD34+CB-MKs and 1 × 106 ASCL-MKs. VEGF expression was detected in CD34+CB-MKs and ASCL-MKs, and ELISA revealed that the amount of VEGF in CD34+CB-MKs was higher than in ASCL-MKs (supplemental Figure 2H). Data of isotype control is shown in supplemental Figure 2I.
Electron microscopic examination of ASCL-MKs revealed typical MK organelles, including lobulated nuclei, granules, mitochondria, and a demarcation membrane system (Figure 2E). Images of ASCL-MKs are shown in supplemental Figure 2J and are similar to those described for CD34+CB-MKs in this study and a previous study.29 These observations suggest that ASCL-MKs were obtained from the ASCL cultured in MKLI media for 8 days.
We then examined gene expression in relation to MK differentiation. Among those genes, p45NF-E2 is one of the determinant transcription factors regulating the differentiation into MK lineages.8 In the present study, gene-expression analysis by quantitative real-time polymerase chain reaction was performed using the ASCL cultured in MKLI media for 0, 2, 4, and 6 days (Figure 3A). We detected increased expression of TPO, p45NF-E2, β-1 tubulin, VWF, and GATA2 during culture. Gene expression of c-MPL, FOG1, Fli1, PF4, RUNX1, and MEIS1 was observed, with a peak at days 0, 2, or 4. On the other hand, gene expression of GATA1 was not detected in the ASCL (day 0) or ASCL-derived cells (days 2, 4, and 6). As a control, we confirmed the experimental conditions for GATA1 using K562 cells (supplemental Figure 3). GATA1 is a key factor for erythroid cell and MK development, whereas GATA2 coordinates MK differentiation in GATA1-deficient and GATA1-mutant cells.43 Thus, the ASCL might have an advantage in MK lineage commitment. These findings indicate that even though ASCL-MKs have intracellular and surface markers normally observed in HSC-derived MKs, ASCL-MKs might have different characteristics from those of HSC-derived MKs. These observations prompted us to use microarray analysis of HSC-derived MKs and ASCL-MKs.
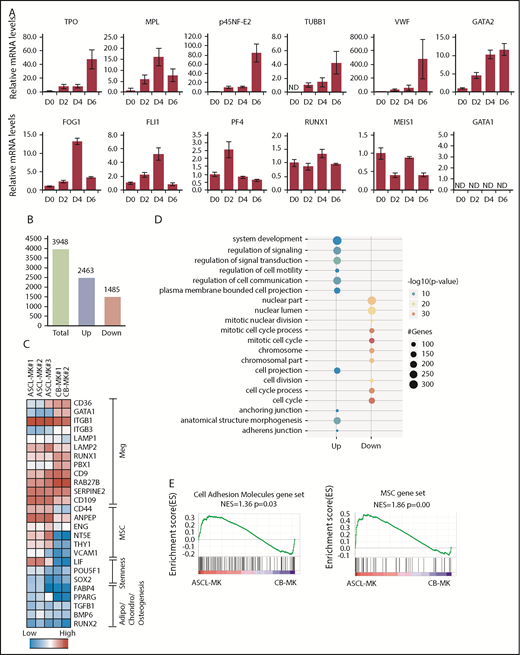
Gene-expression analysis during ASCL-MK differentiation. (A) Expression levels of genes in relation to MK differentiation, as assessed by quantitative real-time PCR. The ASCL was cultured in MKLI media, and the RNA samples were prepared from the cells on days 0, 2, 4, and 6. (B) Number of genes with twofold expression changes in ASCL-MKs compared with CD34+ CB-MKs. (C) Heat map of microarray data showing relative expression of the indicated genes in ASCL-MKs and CD34+CB-MKs. MSC genes were found to be enriched in ASCL-MKs compared with CD34+CB-MKs. Both samples were prepared from the cells on day 8. (D) Top 10 enriched gene-ontology biological processes for ASCL-MKs compared with CD34+CB-MKs. (E) Gene-set enrichment analysis plots showing enrichment for MSC genes (supplemental Figure 5) and cell adhesion genes (gene set name: KEGG_CELL_ADHESION_MOLECULES_CAMS) in ASCL-MKs and CD34+CB-MKs.
Gene-expression analysis during ASCL-MK differentiation. (A) Expression levels of genes in relation to MK differentiation, as assessed by quantitative real-time PCR. The ASCL was cultured in MKLI media, and the RNA samples were prepared from the cells on days 0, 2, 4, and 6. (B) Number of genes with twofold expression changes in ASCL-MKs compared with CD34+ CB-MKs. (C) Heat map of microarray data showing relative expression of the indicated genes in ASCL-MKs and CD34+CB-MKs. MSC genes were found to be enriched in ASCL-MKs compared with CD34+CB-MKs. Both samples were prepared from the cells on day 8. (D) Top 10 enriched gene-ontology biological processes for ASCL-MKs compared with CD34+CB-MKs. (E) Gene-set enrichment analysis plots showing enrichment for MSC genes (supplemental Figure 5) and cell adhesion genes (gene set name: KEGG_CELL_ADHESION_MOLECULES_CAMS) in ASCL-MKs and CD34+CB-MKs.
We performed a microarray study and analyzed the whole-genome expression array data for ASCL-MKs and CD34+CB-MKs. ASCL-MKs expressed MK-related markers and showed high expression levels of mesenchymal markers, such as Cd44, Eng, Nt5e, and Thy1 (known as CD90) (Figure 3C). Gene-ontology analysis identified several significantly enriched biological processes in ASCL-MKs, including anchoring junction and adherens junction (Figure 3D). Gene-set enrichment analysis also revealed that gene expression relevant to cell adhesion (normalized enrichment score = 1.36, P = .03) and mesenchymal markers (normalized enrichment score = 1.72, P = .00) were significantly enriched in ASCL-MKs (Figure 3E; supplemental Figure 4). Taken together, these data suggest that ASCL-MKs showed similar gene expression to HSC-derived MKs and have MSC-related characteristics.
Large-scale manufacture of ASCL-PLTs
The ASCL was cultured in 10 L of MKLI media with a bioreactor (Xuri Cell Expansion System) (supplemental Figure 5A). ASCL-PLTs were characterized by flow cytometric analysis and immunostaining. During ASCL-PLT differentiation from 1 × 108 cells from the ASCL, the number of CD42b+/anucleate platelet-sized cells was 2.7 (± 0.2) × 107 on day 4, 4.2 (± 0.5) × 107 on day 12, and 4.2 (± 0.4) × 107 on day 20 (n = 2; Figure 4A). These data suggest that ASCL-PLTs were obtained for ≥12 days. A similar platelet yield was obtained from the other 2 ASCLs derived from different individuals, and the yield was 11 × 107 and 10 × 107 from 1 × 108 cells from the ASCL (supplemental Figure 5B).
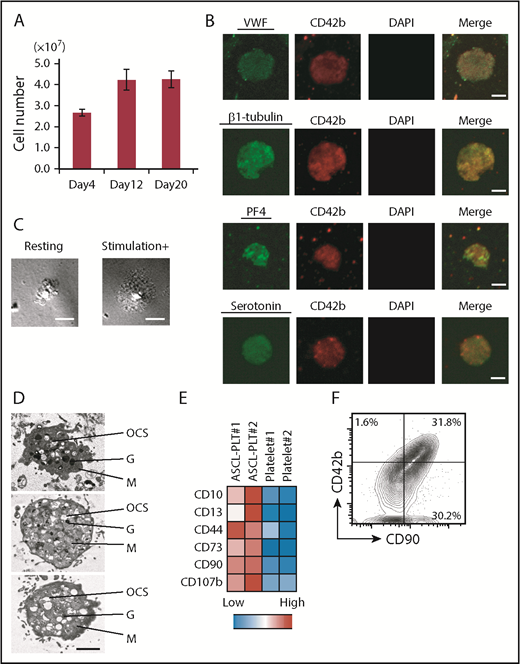
ASCL-PLT production from ASCL-MKs. (A) Number of platelets, as assessed by CD42b+/anucleate platelet-sized cells, produced from 1 × 108 ASCL cells on days 4, 12, and 20. (B) Expression of VWF, β-1 tubulin, PF4, and serotonin in ASCL-PLTs cultured in MKLI media for 12 days. Cells were stained with antibody for CD42b (red), for DAPI (blue), and for VWF, β-1 tubulin, PF4, or serotonin (green). DAPI-stained nuclei were not observed. Scale bars, 2 μm. (C) ASCL-PLTs spreading on fibrinogen-coated glass upon stimulation. Scale bars, 10 μm. (D) Electron microscopic images of produced platelets. Scale bar, 2 μm. (E) Surface marker expression for ASCL-PLTs on day 12 and peripheral platelets. (F) Surface expression of CD42b and CD90 on ASCL-PLTs on day 12. G, granule; M, mitochondrion; OCS, open canalicular system.
ASCL-PLT production from ASCL-MKs. (A) Number of platelets, as assessed by CD42b+/anucleate platelet-sized cells, produced from 1 × 108 ASCL cells on days 4, 12, and 20. (B) Expression of VWF, β-1 tubulin, PF4, and serotonin in ASCL-PLTs cultured in MKLI media for 12 days. Cells were stained with antibody for CD42b (red), for DAPI (blue), and for VWF, β-1 tubulin, PF4, or serotonin (green). DAPI-stained nuclei were not observed. Scale bars, 2 μm. (C) ASCL-PLTs spreading on fibrinogen-coated glass upon stimulation. Scale bars, 10 μm. (D) Electron microscopic images of produced platelets. Scale bar, 2 μm. (E) Surface marker expression for ASCL-PLTs on day 12 and peripheral platelets. (F) Surface expression of CD42b and CD90 on ASCL-PLTs on day 12. G, granule; M, mitochondrion; OCS, open canalicular system.
To examine the expression of intracellular factors in relation to platelet production and function, the expression of VWF, β-1 tubulin, PF4, and serotonin was evaluated by immunostaining. We observed expression of these factors in ASCL-PLTs (Figure 4B; supplemental Figure 5C). We also examined ASCL-PLT staining with anti-serotonin antibody and anti-PF4 antibody (supplemental Figure 5C), as well as CD49b expression in ASCL-PLTs (supplemental Figure 5D). 4′,6-Diamidino-2-phenylindole (DAPI)-stained nuclei were not observed. Data for their isotype control are shown in supplemental Figure 5E. These immunostaining experiments were performed on ASCL-PLTs spread onto fibrinogen-coated glass in the presence of stimulation but not in the absence of stimulation (Figure 4C). Electron microscopic analysis of ASCL-PLTs revealed typical platelet organelles, including an open canalicular system, granules, and mitochondria (Figure 4D), which were similar to those described for HSC-derived platelets.29 Scanning electron microscopy revealed discoid ASCL-PLTs with smooth membranes in the absence of stimulation and ASCL-PLTs with developing tethers in the presence of stimulation (supplemental Figure 5F). Electron microscopic images of induced platelets from CD34+ cord blood cells are also shown (supplemental Figure 5G). These data demonstrate that ASCL-PLTs can be obtained in a large-scale culture system.
Additionally, we examined the expression of surface markers on ASCL-PLTs, because gene-expression analyses indicated that ASCL-MKs have the same gene expression as HSC-MKs and MSCs. ASCL-PLTs showed higher expression of CD10, CD13, CD44, CD73, CD90, and CD107b (Figure 4E). Among them, CD73 and CD90 are cell markers which indicate MSCs according to the criteria by The International Society for Cellular Therapy. As shown in Figure 3E, gene expressions which are related to cell adhesion were significantly enriched in ASCL-MKs, and CD90 is involved in cell adhesion. It is known that cell adhesion has an important role in platelet functions. Thus, we analyzed the expression of CD42b and CD90. More than 95% of CD42+ cells expressed CD90 (Figure 4F). Data for their isotype control are shown in supplemental Figure 5H.
Functional comparison of ASCL-PLTs and platelet concentrates
Platelets have an essential role in hemostasis through platelet activation.44,45 In the steady-state, platelets are not activated and do not aggregate. Platelet activation is caused by stimulation, such as an attachment of exposed subendothelial cells and high-shear blood flow in vivo, and by adding agonists and flow conditions in vitro. We next compared the functionality of ASCL-PLTs with that of platelet concentrates obtained from the Japanese Red Cross Society. Stimulation with thrombin (1 U/mL), epinephrine (1 μg/mL), calcium (1.5 mM), and magnesium (2 mM) increased binding of Alexa Fluor 488–labeled fibrinogen, a ligand for activated platelets, to ASCL-PLTs in a similar manner to platelet concentrates (Figure 5A). The stimulation increased PAC1 binding, a marker for platelet activation, to ASCL-PLTs to a greater extent than to the platelet concentrates (Figure 5A). Surface exposure of P-selectin, another marker of platelet activation, tended to be higher on ASCL-PLTs than on platelet concentrates (Figure 5A). ASCL-PLTs derived from different individuals did not differ with regard to fibrinogen binding, PAC1 binding, or P-selectin surface exposure (supplemental Figure 6A). P-selectin expression at baseline (ie, no stimulation in ASCL-PLTs and platelet concentrates) was assessed by mean fluorescence, and the baseline for ASCL-PLTs was higher than that for platelet concentrates (supplemental Figure 6B). Agonist-induced aggregation was observed in ASCL-PLTs and platelet concentrates (Figure 5B). Adenosine 5′-diphosphate (ADP) (20 μM), collagen (5 μg/mL), ristocetin (1.2 mg/mL), and epinephrine (10 μM) were used as agonists. Although the maximum aggregation induced by ADP or ristocetin in ASCL-PLTs was higher than that in platelet concentrates, the maximum epinephrine-induced aggregation of ASCL-PLTs was lower than that in platelet concentrates. The maximum aggregation induced by collagen was similar between ASCL-PLTs and platelet concentrates. No spontaneous aggregation was observed in ASCL-PLTs or platelet concentrates in these aggregation studies. We examined platelet contents using 2 × 107 cells. Thromboxane B2 secretion levels under calcium (10 mM) stimulation in ASCL-PLTs (61.9 ± 1.8 ng/mL) were lower than those in platelet concentrates (2250.2 ± 56.5 ng/mL), and the levels were decreased in the presence of 100 μM aspirin for 15 minutes: 8.1 ± 3.1 ng/mL and 436.4 ± 75.6 ng/mL, respectively. PF4 levels were 1.1 ± 1.7 pg for ASCL-PLTs and 3.1 ± 0.4 pg for platelets, and VEGF levels were 44.5 ± 7.2 pg for ASCL-PLTs and 78.4 ± 4.0 pg for platelets.
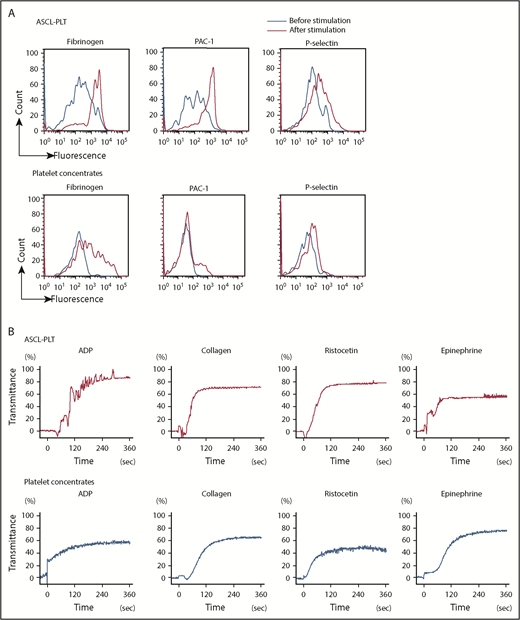
Functional assay for ASCL-PLTs and platelet concentrates. (A) Fibrinogen binding, PAC1 binding, and surface exposure of P-selectin for ASCL-PLTs and platelet concentrates in the presence or absence of stimulation (thrombin, 1 U/mL; epinephrine, 1 μg/mL; calcium, 1.5 mM; and magnesium, 2 mM). (B) Agonist-induced aggregation of ASCL-PLTs and platelet concentrates recorded under light transmission aggregometry. ADP (20 μM), collagen (5 μg/mL), ristocetin (1.2 mg/mL), and epinephrine (10 μM) were used as agonists.
Functional assay for ASCL-PLTs and platelet concentrates. (A) Fibrinogen binding, PAC1 binding, and surface exposure of P-selectin for ASCL-PLTs and platelet concentrates in the presence or absence of stimulation (thrombin, 1 U/mL; epinephrine, 1 μg/mL; calcium, 1.5 mM; and magnesium, 2 mM). (B) Agonist-induced aggregation of ASCL-PLTs and platelet concentrates recorded under light transmission aggregometry. ADP (20 μM), collagen (5 μg/mL), ristocetin (1.2 mg/mL), and epinephrine (10 μM) were used as agonists.
We then investigated the in vivo kinetics of ASCL-PLTs and platelet concentrates by infusing these cells into irradiated immunodeficient NSG mice (2.0 Gy, 7 days) (Figure 6A). Blood samples from recipient mice were analyzed for the presence of human platelets at 0 minutes, 30 minutes, 2 hours, 4 hours, 6 hours, and 24 hours after infusion. The kinetics of ASCL-PLTs was similar to that of the platelet concentrates, with a peak at 30 minutes. The presence of human platelets at 2 hours after infusion in the ASCL-PLT group was higher than that in the platelet concentrate group, although the percentage varied according to sample (n = 3; Figure 6B).
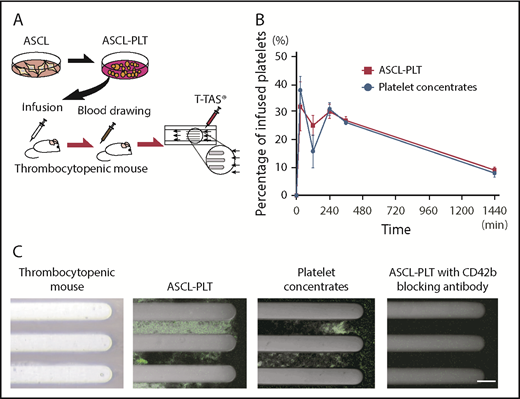
Infusion study for kinetics and incorporation into thrombus formation under flow conditions. (A) Schema of the human platelet infusion study using thrombocytopenic NSG mice. (B) The presence of human platelets, as assessed by CD42b+/anucleate platelet-sized cells, in mouse blood after the infusion of ASCL-PLTs or platelet concentrates. (C) Ex vivo thrombus formation under flow conditions, observed under a fluorescence microscope. The blood samples were taken from thrombocytopenic NSG mice infused with ASCL-PLTs or platelet concentrates. Scale bar, 50 μm.
Infusion study for kinetics and incorporation into thrombus formation under flow conditions. (A) Schema of the human platelet infusion study using thrombocytopenic NSG mice. (B) The presence of human platelets, as assessed by CD42b+/anucleate platelet-sized cells, in mouse blood after the infusion of ASCL-PLTs or platelet concentrates. (C) Ex vivo thrombus formation under flow conditions, observed under a fluorescence microscope. The blood samples were taken from thrombocytopenic NSG mice infused with ASCL-PLTs or platelet concentrates. Scale bar, 50 μm.
Using the blood samples from recipient mice 24 hours after infusion, we tested thrombus formation under flow conditions, because blood flow is a key factor for thrombus formation in vivo. We used a Total Thrombus-formation Analysis System, an automated microchip flow chamber system.46,47 Blood samples labeled with anti-CD41 antibody (clone SZ22) were perfused on a microchip coated with collagen, a major subendothelial matrix. Incorporation of cells expressing human CD41 into the thrombi on the collagen surface was observed in samples from ASCL-PLT–infused mice and platelet concentrate–infused mice (Figure 6C). Thrombi were not observed in whole blood samples from irradiated thrombocytopenic NSG mice without an infusion of ASCL-PLTs or platelet concentrates (Figure 6C). To examine whether ASCL-PLTs or platelet concentrates are passively incorporated into thrombi, a perfusion study was performed in the presence of the anti-human CD42b blocking antibody HIP1. Thrombi were slightly formed on the collagen surface in the blood samples treated with HIP1 (Figure 6C). These findings indicate that manufactured ASCL-PLTs have in vivo kinetics and functions similar to those of platelet concentrates obtained from the Japanese Red Cross Society.
Discussion
The present study revealed the characterization of the ASCL, ASCL-MKs, and ASCL-PLTs, and we compared platelet functions and in vivo kinetics between ASCL-PLTs and platelet concentrates obtained from the Japanese Red Cross Society. The ASCL could expand under long-term culture with genetic stability. We also assessed the genetic stability by next-generation sequence analysis (data not shown). As we reported previously, an adipogenic cell line is able to differentiate into MKs whereas a nonadipogenic cell line is not. Thus, as a result of its heterogeneity, the proliferation and MK differentiation ability of ASCs vary considerably. In contrast, the ASCL ameliorates this concern, because this population remains relatively homogeneous; only adipogenic cells among ASCs are selected through the establishment process. The ASCL fulfilled the minimal criteria for MSCs established by The International Society for Cellular Therapy.32,33 Clinical applications of ASC/MSC-derived chondrocytes and osteoblasts are under intensive investigation.48 Bone marrow MSCs have been used for patients with graft-versus-host disease, an immune-related disease. The ASCL is a candidate cell source for the manufacture of donor-independent platelets, because it can differentiate into MK lineages using endogenous genes and the cytokine TPO. The large-scale manufacture of ASCL-PLTs was achieved using a simple method. Although MKs and subsequent platelets are hematopoietic cells, distinct from mesenchymal cells, the ASCL and ASCs contain critical factors that drive MK lineages.
Platelets play critical roles in hemostatic plug formation, which is mediated by platelet activation. Platelets in the steady-state are not activated and cannot trigger hemostatic plug formation. Platelet activation is caused by interactions of platelet membrane receptors, such as glycoprotein (GP)IIb/IIIa (also known as CD41/CD61) and GPIb/IX/V (also known as CD42b/CD42c/CD42a/CD42d), with their ligands. The importance of these receptors in platelet functions is illustrated in patients with a deficiency or dysfunction of these receptors (eg, Glanzmann thrombasthenia and Bernard-Soulier syndrome, respectively).49-51 We assessed the expression and function of these major receptors using well-established methods. ASCL-MKs and ASCL-PLTs have similar characteristics to those of CD34+CB-MKs and peripheral platelets, and microarray analysis and flow cytometry analysis revealed that ASCL-MKs and ASCL-PLTs have additional characteristics normally observed in MSCs. ASCL-PLTs have functions that are unique from, as well as similar to, those of peripheral platelets. Compared with peripheral platelets, ASCL-PLTs exhibit higher levels of PAC1 binding, P-selectin surface exposure, ADP-induced platelet aggregation mediated by the ADP receptor, ristocetin-induced platelet aggregation mediated by GPIb-α, and ASCL-PLT levels in mouse blood 2 hours after infusion. ASCL-PLTs and peripheral platelets showed similar levels of fibrinogen binding, collagen-induced platelet aggregation mediated by GPIa/IIa and GPVI, and the similar kinetics in mouse blood at 30 minutes, 4 hours, 6 hours, and 24 hours after infusion. Compared with peripheral platelets, ASCL-PLTs have lower thromboxane B2 levels in the presence of stimulation and lower epinephrine-induced platelet aggregation mediated by the epinephrine receptor. Comparing to platelet concentrates, some of ASCL-PLTs are in an active state at baseline according to the results of P-selectin exposure, and this is often seen in pathological diseases, including malignancy, diabetes, and autoimmune disease. Although no spontaneous aggregation before stimulation was observed in an agonist-induced platelet aggregation study, there is the possibility that ASCL-PLTs could lead to thrombotic complications. One way to reduce the hyperactivity of ASCL-PLTs for infusion may be to decrease the number of cells infused. Also, we are studying a monitoring system to optimize the use of ASCL-PLTs for infusion. We do not have direct evidence to support our idea; thus, it is important to perform further studies to optimize ASCL-PLTs as candidates for clinical applications.
Platelet yields are critical for establishing an ideal system for manufacturing platelets. More than 1011 platelets are used per transfusion in an adult patient. MKs proliferate little or not at all without gene transduction; thus, the development of an expandable cell source is critical for platelet manufacture. Human iPS cells are considered a candidate source for platelet manufacture. Nakamura et al reported that a single MK obtained from iPS cells transfected with c-MYC, BMI1, and BCL-XL released 3 to 10 platelets.16 Feng et al reported the generation of universal platelets from iPS cells with knockout of the b2-microglobulin gene and demonstrated that a single MK progenitor generated ∼6 platelets.22 Moreau et al achieved a chemically defined large-scale production of MKs from iPS cells, with a single MK releasing ∼5 platelets.23 At present, a single cell from the ASCL generates ≥5 to 10 ASCL-PLTs in a culture system using 10 L of medium in just 12 days. Although >95% of the CD42+ cells expressed CD90, the frequency of CD90+ cells was 60% of the filtered cells. Thus, ASCL-PLTs do not represent a completely pure population. With regard to contamination with MSCs or ASCL-MKs, <2% of the filtered cells were nucleated cells, as assessed by flow cytometry with 7-aminoactinomycin D (K.T., Y.O.-U., and Y.M., unpublished data), suggesting that >98% of the filtered cells were not MSCs or ASCL-MKs.
We also observed that the CD41− cells cultured in MKLI media did not have a high-ploidy population. CD41+ cells had a high-ploidy population. Also, most of the CD41− cells were dying in serum-free culture with MKLI media. Although it remains unclear whether CD41/CD42b double-positive MKs release platelets, a high-ploidy population in CD41+/CD42b− ASCL-MKs released platelets in a pilot study (K.T., Y.O.-U., and Y.M., unpublished data), and it is possible that CD41+ MKs release platelets.
Differentiation of cord blood cells into platelets is more efficient than for the ASCL into ASCL-PLTs. A possible reason for this discrepancy is as follows. The present method does not require gene transfer and utilizes endogenous TPO. Also, the ASCL differentiates into MKs and platelets, as well as into chondrocytes, adipocytes, and osteoblasts. Although we do not have direct evidence to explain why only 63% of cells became MKs, it is possible that there are MK-primed cells among the ASCL population. MSC markers were observed in >95% of cells in the ASCL (Figure 1D), but expression patterns are heterogeneous (eg, CD71 expression). Thus, further studies are necessary to identify MK-primed ASCLs to better understand the efficiency of ASCL-PLT transformation.
In summary, we report the characteristics of an expandable ASCL from subcutaneous adipose tissues and the manufacture of functional ASCL-PLTs in a large-scale culture. Neither the establishment of the ASCL nor its differentiation into ASCL-MKs and subsequent ASCL-PLTs requires gene transfer, and endogenous TPO is used for differentiation. The present protocol is a simple method, further enhancing the clinical application of our approach.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Kazuya Hosokawa and Tomoko Ohnishi (Research Institute, Fujimori Kogyo) for critical advice about the perfusion study and Noriko Takizawa and Aya Shimodaira for technical assistance.
This work was supported by Grants-in Aid from the Japan Agency for Medical Research and Development (Translational Research Network Program [Y.M. and T.M.]), the Kanagawa Academy of Science and Technology (Y.M.), and Japan Society for the Promotion of Science KAKENHI (grants 25462800 and 16K11379 [M.Y., T.M., and Y.M.] and grant 26461410 [Y.M.]).
Authorship
Contribution: K.T. and Y.O.-U. performed the studies, analyzed data, and prepared the manuscript; M.Y. prepared a sample from adipose tissues and interpreted data; T.M. performed the genetic study and analyzed and interpreted data; M.M and S.O. provided critical suggestions; Y.I. provided advise about the study design, interpreted data, and prepared the manuscript; and Y.M. designed and supervised the study and prepared the manuscript.
Conflict-of-interest disclosure: Y.M. receives research support from Nissui Pharmaceutical, Tosoh, AdipoSeeds, and Fujimori Kogyo. Y.M., Y.I., and Y.O.-U. own shares in AdipoSeeds and are inventors on patent PCT/JP2014/003445. The remaining authors declare no competing financial interests.
Correspondence: Yumiko Matsubara, Clinical and Translational Research Center, Keio University School of Medicine, 35 Shinanomachi, Shinjuku-ku, Tokyo 160-8582, Japan; e-mail: yumikoma@keio.jp.