TO THE EDITOR:
Non-Hodgkin B-cell lymphomas are almost invariably derived from B lymphocytes that have undergone productive V(D)J rearrangements of their immunoglobulin genes and may additionally have experienced somatic hypermutation. In the case of follicular lymphomas, malignant clones undergo ongoing somatic hypermutation of the rearranged V(D)J allele as they further expand. The resulting B-cell receptor idiotype contains novel, tumor-specific sequences and thus represents a class of neoantigen unique to B-cell malignancies. Major histocompatibility complex (MHC) presentation and T-cell recognition of the B-cell idiotype have been demonstrated in mice1,2 and in humans.3-7
Presentation of B-cell idiotype has typically been inferred through activation of cognate T cells. Immunoglobulin-derived peptides have been eluted from class II MHC (MHC-II) in a murine B-cell hybridoma line.8 Mass spectrometry has greatly improved direct detection of B-cell MHC ligands9 including germline-encoded immunoglobulin peptides. However, prior studies have found that germline-derived immunoglobulin peptides are not effective targets for T cells due to immune tolerance.10-12 Thus, immunoglobulin neoantigens may be the most critical antigens for idiotype recognition, but their detection remains a challenge due to their personalized nature.
We recently used a novel strategy of antigen-presentation profiling in mantle cell lymphoma (MCL) through integrated tumor genomic profiling and proteomic characterization of tumor-derived class I MHC (MHC-I) and MHC-II ligands by immunoprecipitation and liquid chromatography (LC)–tandem mass spectrometry (MS/MS).13 Within this analysis, we included the sequencing of the rearranged lymphoma immunoglobulin alleles to directly identify immunoglobulin neoantigens derived from unique V(D)J rearrangements and somatic mutations.13 We found MCL neoantigens frequently presented by MHC-II, but only rarely observed presentation of variable region peptides by MHC-I. However, it is unknown whether this pattern of biased immunoglobulin presentation is shared by other B-cell lymphoma subtypes. Here, we profile the antigens associated with MHC-I and MHC-II of follicular lymphoma (FL), diffuse large B-cell lymphoma (DLBCL), and chronic lymphocytic leukemia (CLL) and establish that immunoglobulin-derived neoantigen presentation by MHC is a general phenomenon of B-cell malignancies.
We performed MHC-I and MHC-II antigen-presentation profiling of 6 FL, 1 DLBCL, and 2 CLL samples. To enhance discovery of personalized antigens, including immunoglobulin neoantigens, we sequenced the lymphoma immunoglobulin heavy-chain genes from all samples and light-chain sequences for 5 of the FL patients with sufficient material available. All specimens were obtained with informed consent in accordance with the Declaration of Helsinki, and this study was approved by Stanford University’s Administrative Panels on Human Subjects in Medical Research.
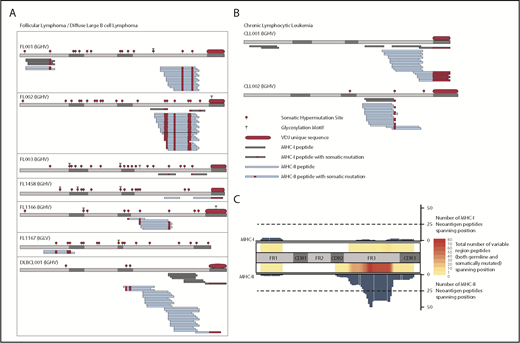
We identified a total of 11 immunoglobulin-derived neoantigens presented by MHC-I from 4 of 7 FL/DLBCL tumors (Figure 1A) and both CLL samples (Figure 1B). MHC-II presentation of immunoglobulin neoantigens was observed in all patients studied, with a total of 70 discovered class II neoantigens. Consistent with our prior observations in MCL, the presentation of the immunoglobulin variable region was also strongly biased toward MHC-II in these FL, DLBCL, and CLL cases (Figure 1C; supplemental Figure 1, available on the Blood Web site). The most frequently presented region of the immunoglobulin heavy chain was the framework 3 (FR3) region, similar to the pattern seen in MCL. We observed few immunoglobulin-derived MHC-I ligands. These ligands corresponded to active areas of MHC-II presentation, again consistent with the hotspot pattern of antigen presentation seen among melanoma MHC ligands.14 We observed NX(S/T) glycosylation motifs created through somatic hypermutation or VDJ recombination in all 6 FL tumors, but never in other subtypes. Because our method did not directly target glycosylated peptides, we cannot conclude whether glycosylated peptides are presented.
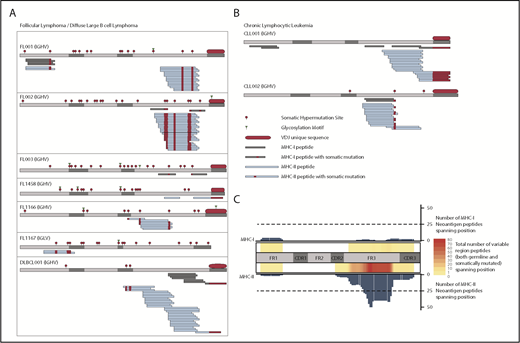
Presentation of lymphoma immunoglobulin peptides by MHC. FL/DLBCL (A) and CLL (B) specimens were lysed, and MHC-I and MHC-II were immunoprecipitated in parallel. MHC-bound peptides were acid-eluted, fractionated by LC, and analyzed by MS. MHC-I (gray) and MHC-II (blue) bound peptides are depicted. Somatically mutated residues (either through somatic hypermutation or VDJ rearrangement) are shown in red. (C) Heatmap depicting the number of total class I (top) and class II (bottom) ligands across the immunoglobulin heavy chain for all patients. Both germline-derived and neoantigen peptides are included. The bar plots represent the number of neoantigen peptides spanning each position of the heavy chain for class I (top) and class II (bottom). CDR, complementarity-determining region; FR, framework region; IGHV, immunoglobulin heavy-chain variable region; IGLV, immunoglobulin light-chain variable region.
Presentation of lymphoma immunoglobulin peptides by MHC. FL/DLBCL (A) and CLL (B) specimens were lysed, and MHC-I and MHC-II were immunoprecipitated in parallel. MHC-bound peptides were acid-eluted, fractionated by LC, and analyzed by MS. MHC-I (gray) and MHC-II (blue) bound peptides are depicted. Somatically mutated residues (either through somatic hypermutation or VDJ rearrangement) are shown in red. (C) Heatmap depicting the number of total class I (top) and class II (bottom) ligands across the immunoglobulin heavy chain for all patients. Both germline-derived and neoantigen peptides are included. The bar plots represent the number of neoantigen peptides spanning each position of the heavy chain for class I (top) and class II (bottom). CDR, complementarity-determining region; FR, framework region; IGHV, immunoglobulin heavy-chain variable region; IGLV, immunoglobulin light-chain variable region.
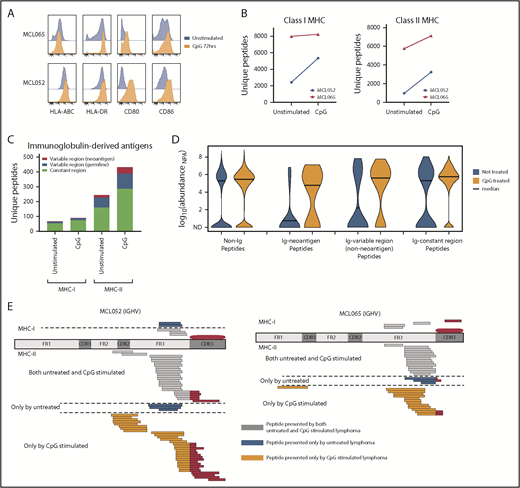
We next sought to determine whether we could increase immunoglobulin neoantigen presentation by activating B-cell lymphomas with a Toll-like receptor 9 (TLR9) agonist to promote MHC-II presentation.15 Activation of MCL increased MHC-II expression with little effect on MHC-I expression (Figure 2A). However, we found that global recovery of both MHC-I and MHC-II ligands was enhanced by TLR9 stimulation (Figure 2B). These included immunoglobulin-derived neoantigens presented by MHC-II, but not MHC-I (Figure 2C-E). Although TLR9 stimulation does have the potential to promote growth of malignant B cells, TLR9 agonists have been safely and effectively used in clinical trials of low-grade B-cell lymphomas and therefore represent a potential strategy to enhance immunoglobulin neoantigen presentation.16,17
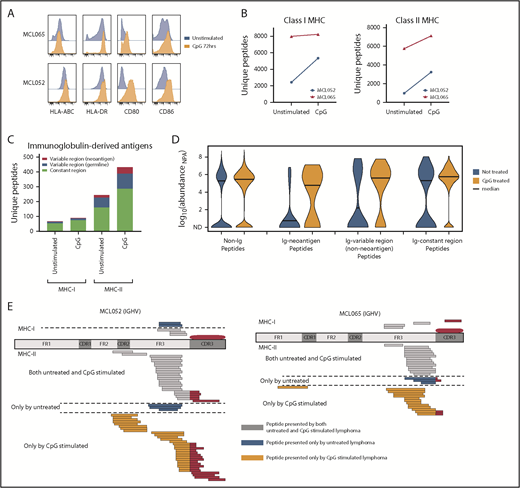
Activation of MCL with a TLR9 agonist alters MHC presentation of the lymphoma immunoglobulin. Equal numbers of MCL cells were incubated for 72 hours with or without cytosine guanine dinucleotide (CpG), a TLR9 agonist. (A) Flow cytometry assessment of antigen-presentation molecule expression with (orange) and without (blue) CpG treatment. Two representative cases of MCL are shown. (B) MHC-I– and MHC-II–bound ligands were purified and analyzed by LC and MS. The total number of unique MHC-I (left) and MHC-II (right) associated peptides is shown with and without CpG treatment. (C) The number of unique MHC-I– and MHC-II–bound ligands derived from the lymphoma immunoglobulin (Ig) constant and variable regions is shown. (D) Kernel density violin plots of the estimated normalized abundance of MHC-II–bound peptides recovered from unstimulated and CpG-treated MCL. (E) Mapping of MHC-I and MHC-II presentation of the MCL immunoglobulin is shown. MHC ligands recovered from CpG-treated cells and untreated cells are separated by a dashed line. ND, not detected; NPA, normalized protein abundance.
Activation of MCL with a TLR9 agonist alters MHC presentation of the lymphoma immunoglobulin. Equal numbers of MCL cells were incubated for 72 hours with or without cytosine guanine dinucleotide (CpG), a TLR9 agonist. (A) Flow cytometry assessment of antigen-presentation molecule expression with (orange) and without (blue) CpG treatment. Two representative cases of MCL are shown. (B) MHC-I– and MHC-II–bound ligands were purified and analyzed by LC and MS. The total number of unique MHC-I (left) and MHC-II (right) associated peptides is shown with and without CpG treatment. (C) The number of unique MHC-I– and MHC-II–bound ligands derived from the lymphoma immunoglobulin (Ig) constant and variable regions is shown. (D) Kernel density violin plots of the estimated normalized abundance of MHC-II–bound peptides recovered from unstimulated and CpG-treated MCL. (E) Mapping of MHC-I and MHC-II presentation of the MCL immunoglobulin is shown. MHC ligands recovered from CpG-treated cells and untreated cells are separated by a dashed line. ND, not detected; NPA, normalized protein abundance.
Our data support the strong bias of MHC-II presentation of immunoglobulin peptides across B-cell lymphoma subtypes. Although this bias applies to the entire immunoglobulin molecule, it is more prominent in the presentation of the variable regions (supplemental Figure 1). We observed a consistent predisposition toward presenting the FR3 region of the immunoglobulin heavy chain. Selective presentation of framework regions was previously suggested for MHC-I based on computational predictions, and cytotoxic T-cell lymphocyte responses against framework-derived antigens have been observed.4,18 In the case of MHC-II, prior computational studies have predicted MHC-II presentation to be limited to specific regions of the immunoglobulin variable region with high affinity for MHC.19 This is consistent with the hotspot of presentation in our study, although we find only a single hotspot in FR3, and not the multiple hotspots as predicted by MHC-II–binding algorithms. Similarly to MCL,13 we did observe occasional MHC ligands centered on the complementarity-determining region 3 (CDR3). CDR3 has been predicted to be an actively presented immunoglobulin region based on the presence of frequent flanking cathepsin cleavage sites.20 However, we note that our identified MHC-II ligands were not enriched for the previously noted cathepsin cleavage signature. Taken together, the presentation pattern we observed suggests that the FR3 and CDR3 regions may be the portion of lymphoma immunoglobulin most amenable to immunotherapy targeting.
Preferential MHC-II presentation of the immunoglobulin variable region suggests that CD4-mediated immune strategies may be more effective in treating B-cell malignancies. Our findings are consistent with prior reports of lymphoma immunoglobulin recognition by CD4+ T cells.6,7,13 T cells targeting immunoglobulin variable regions have been shown to be capable of both preventing and treating B-cell malignancies in mice.21,22 Upon ex vivo expansion, human immunoglobulin neoantigen-targeting CD4+ T cells produce interferon γ and may become capable of directly killing autologous lymphoma cells.7,13
Conversely, the abundant presentation of lymphoma immunoglobulin neoantigens by MHC-II may also reflect a role in promoting lymphoma pathogenesis. For example, CD4 T-cell recognition of B-cell immunoglobulin sequences can contribute to lymphomagenesis in murine models23 and in CLL.6 Thus, the phenotype of neoantigen-specific CD4 T cells may be a critical consideration in designing immune strategies targeting lymphoma immunoglobulin. One potential strategy would be to promote T-helper type 1 differentiation of neoantigen-specific CD4 T cells. Immunoglobulin-targeting T-helper type 1 cells appear more cytotoxic in treating murine B-cell lymphoma22 and could perhaps be generated through targeting of interleukin-2–inducible T-cell kinase by either interleukin-2–inducible T-cell kinase inhibitors or certain Bruton tyrosine kinase inhibitors such as ibrutinib.24
We performed whole-exome sequencing of 3 FL samples to detect presentation of nonimmunoglobulin, nonsynonymous somatic mutation-bearing neoantigens. We found no examples of MHC presentation of nonimmunoglobulin neoantigens. This contrasts with similar antigen-profiling efforts in high-mutation-burden diseases such as melanoma where neoantigens are found among MHC ligands.14,25 We also did not find shared lymphoma antigens, although our study was not designed to identify such antigens in contrast to previous work in CLL and multiple myeloma.26,27
Our direct antigen-presentation profiling confirms that lymphoma immunoglobulin is a commonly presented B-cell lymphoma neoantigen. Three prior phase 3 trials of idiotype vaccine have shown limited efficacy,28-30 with only 1 trial showing improved disease-free survival.29 Nevertheless, our data suggest that the lymphoma immunoglobulin deserves continued consideration as an immunotherapy target in the current era of neoantigen-targeting immunotherapies. Activation of antigen presentation by TLR agonists represents a strategy to augment anti-immunoglobulin immunotherapy.
The online version of this article contains a data supplement.
Acknowledgments
The authors are grateful to the patients who participated in this study. The authors also thank Shoshana Levy and Max Diehn for their critical feedback, as well as Etelka Gabriel and Cynthia Glover for their efforts in collecting samples.
This work was supported by grants from the National Institutes of Health, National Cancer Institute (U01 CA194389 [M.M.D., R.L., J.E.E., and A.A.A.], Program Project Grant CA49605 [R.L.], R35 CA197353 [R.L.], and K08 CA207882-01A1 [M.S.K.]); an American Society of Hematology Scholar Award (A.A.A.); a grant from the V-Foundation (A.A.A.); and a grant from the Damon Runyon Cancer Research Foundation (A.A.A.). J.E.E. is a Damon Runyon-Rachleff Innovation Awardee, and recipient of a W. M. Keck Foundation Medical Research Grant. M.S.K. received a Conquer Cancer Foundation Young Investigator Award and a Fellow Award from the Leukemia & Lymphoma Society. N.O. received a Knut and Alice Wallenberg Foundation Postdoctoral Fellowship. Additional support was received from National Institutes of Health, National Institute of General Medical Sciences grant S10 RR02933801.
Authorship
Contribution: M.S.K., N.O., R.L., J.E.E., and A.A.A. conceived and designed the research; M.S.K., B.S., R.L., and A.A.A. provided patient samples and clinical data; M.S.K., N.O., B.C., B.S., T.S., C.L.L., L.Z., D.K.C., M.M.D., R.L., J.E.E., and A.A.A. performed the research and/or analyzed data; M.S.K., N.O., R.L., J.E.E., and A.A.A. wrote the paper; and all authors critically reviewed and edited the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ash A. Alizadeh, School of Medicine, Stanford University, 259 Campus Dr, Stanford, CA 94305; e-mail: arasha@stanford.edu; and Joshua E. Elias, Stanford University, 318 Campus Dr, Stanford, CA 94305; e-mail: josh.elias@stanford.edu.