Key Points
Substantive familial risks are associated with each hematological malignancy, younger diagnosis age, and multiple affected relatives.
The familial relative risks provide evidence for shared etiology between the specific hematological malignancies.

Abstract
Estimating familial cancer risks is clinically important in being able to discriminate between individuals in the population at differing risk for malignancy. To gain insight into the familial risk for the different hematological malignancies and their possible inter-relationship, we analyzed data on more than 16 million individuals from the Swedish Family-Cancer Database. After identifying 153 115 patients diagnosed with a primary hematological malignancy, we quantified familial relative risks (FRRs) by calculating standardized incident ratios (SIRs) in 391 131 of their first-degree relatives. The majority of hematological malignancies showed increased FRRs for the same tumor type, with the highest FRRs being observed for mixed cellularity Hodgkin lymphoma (SIR, 16.7), lymphoplasmacytic lymphoma (SIR, 15.8), and mantle cell lymphoma (SIR, 13.3). There was evidence for pleiotropic relationships; notably, chronic lymphocytic leukemia was associated with an elevated familial risk for other B-cell tumors and myeloproliferative neoplasms. Collectively, these data provide evidence for shared etiological factors for many hematological malignancies and provide information for identifying individuals at increased risk, as well as informing future gene discovery initiatives.
Introduction
Each of the hematological malignancies is characterized by a distinctive clinical phenotype reflective of differences in its progenitor cell of origin and underling biology. The lymphomas, B- and T-cell leukemias, and myeloma are of lymphoid origin, arising at differing stages of maturation.1 Acute and chronic myeloid leukemia (CML), myelodysplastic syndrome (MDS), and the myeloproliferative diseases are all derived from a myeloid progenitor.2 Although many hematological malignancies are individually rare, collectively they contribute significantly to the overall cancer burden in the population.3
Aside from exposure to DNA-damaging agents and the association between Epstein-Barr virus (EBV), HIV, human T-cell lymphotropic virus (HTLV), and Helicobacter pylori with specific lymphoma subtypes, the etiological basis of most hematological malignancies is poorly understood.4,,,,,,,,,-14
Epidemiological observational studies and reports of families segregating hematological malignancies over the years have supported the role of inherited factors in disease etiology.15,,-18 Direct evidence for predisposition to hematological tumors is provided by the increased risk associated with a number of rare inherited syndromes19 (eg, Fanconi anemia,20 Diamond-Blackfan anemia,21 and dyskeratosis congenita22 ), as well as rare germline mutations in a number of genes causing Mendelian susceptibility (ANKRD26,23 CEBPA,24 DDX41,25 ELANE,26 ETV6,27 GATA2,28 HAX1,29 RUNX1,30 SAMD9,31 SAMD9L,32 SRP72,33 and LSD134 ). Recently, genome-wide association studies have provided evidence for a heritable basis to sporadic forms of acute lymphoblastic leukemia (ALL),35 Hodgkin lymphoma (HL),36,37 diffuse large B-cell lymphoma (DLBCL),38 primary central nervous system lymphoma,39 follicular lymphoma (FL),40 marginal zone lymphoma,41 lymphoplasmacytic lymphoma/Waldenström macroglobulinemia (LPL/WM),42 chronic lymphocytic leukemia (CLL),43 multiple myeloma (MM),44 and the myeloproliferative neoplasms (MPNs).45
The increasing importance of recognizing inherited predisposition to hematological malignancy is underscored by the 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia, which has made recognition of a familial disease a component of diagnosing leukemia.2 Aside from precise estimates of familial risks for the hematological malignancies being clinically important in being able to discriminate between individuals at differing risk,46,47 such information is relevant to understanding the nature of genetic susceptibility (ie, contextualizing the effect of known syndromic forms of predisposition).19,48,49
Previous studies of familial risk have reported high disease-specific familial risks for ALL,17 HL,16 DLBCL,50 FL,50 MCL,15 LPL/WM,51 CLL,52 MM,53 polycythemia vera (PV),54 and essential thrombocythemia (ET).54 Furthermore, an increased risk for lymphoproliferative disorders has been reported in the relatives of patients with LPL/WM,51 DLBCL,15 FL,15 small lymphocytic lymphoma,15 CLL,52 and MM.55 However, these studies have limited power to quantify subtype-specific familial risks and to dissect familial risk based on age, sex, and familial relationship. In addition, these studies have not explored familial associations across the complete spectrum of hematological malignancies.
By analyzing the Swedish Family-Cancer Database, we have recently enumerated the familial risks for myeloid malignancies.18 To provide a more comprehensive analysis of the spectrum of all hematological malignancies, we have now extended our analysis of more than 16 million individuals to estimate risk for all lymphoid malignancies and define their inter-relationship. Our analysis provides evidence for shared familial risks consistent with a common etiological basis for a number of the hematological malignancies both within and extending across cell lineage of origin.
Materials and methods
Swedish Family-Cancer Database
We used the Swedish Family-Cancer Database to estimate familial relative risks (FRRs) of the major hematological malignancies. The Swedish Family-Cancer Database was created by linking information from the Multi-Generation Register, national censuses, the Swedish Cancer Registry, and death notifications.56 The Swedish Cancer Registry, established in 1958, is based on the compulsory reporting of all cancer diagnoses, providing near-complete coverage of all cancer registrations in Sweden.57 The 2015 update of these data includes a total population of more than 16.1 million individuals. The Swedish Family-Cancer Database is composed of all families from the Multigeneration Register since its inception.58 Since the Swedish Cancer Registry started in 1958, some individuals in the first generation will inevitably have been diagnosed with cancer before the registration was initiated. The possible influence of this left truncation on FRR estimation has, however, been analyzed and found not to cause bias.59
We considered all incident cases of hematological malignancies diagnosed between 1958 and 2015. Specifically, we analyzed all incident cases of all hematological malignancies, all myeloid malignancies, the MPNs, PV, ET, myelofibrosis, MPN not otherwise specified (MPN-NOS), CML, MDS, acute myeloid leukemia (AML), ALL, HL, nodular sclerosis HL, mixed cellularity HL, non-HL (NHL), DLBCL, FL, LPL/WM, mantle cell lymphoma (MCL), marginal zone lymphoma, Burkitt lymphoma, small lymphocytic lymphoma, hairy cell leukemia, CLL, MM, mature T-cell lymphoma, anaplastic T-cell lymphomas, and cutaneous T-cell lymphoma. The Swedish Cancer Register has implemented the International Classification of Disease (ICD)-7 since 1958, ICD-9 since 1987, ICD-O/2 and Systematized Nomenclature of Medicine (SNOMED) histopathological codes since 1993, and ICD-O/3 since 2005. We used a combination of ICD-7, ICD-O/2, and SNOMED codes to establish the diagnosis for most of the hematological malignancies. As MDS and subtypes of NHL and HL require ICD-O/2 and SNOMED codes, our analysis for these diseases was confined to data collected from 1993. Because SNOMED codes are based on the Kiel and Rappaport classifications, it was not possible to define all NHL subtypes on the basis of World Health Organization classification. The World Health Organization does, however, provide synonymous definitions across classifications, and these translations were used where possible. Individuals diagnosed with MDS before a diagnosis of AML only contributed the MDS analysis to mitigate against any possible confounding from treatment.
The study was undertaken with approval from the ethics committee at Lund University, Sweden, and was conducted in accordance with the tenets of the Declaration of Helsinki.
Statistical analysis
A Poisson distribution was used to calculate the 95% confidence intervals (CIs). Tests for trend in SIRs were performed by evaluating the likelihood function in collapsed person-time additive Poisson regression models with and without the inclusion of the variable. In the stratified analyses where an FDR appears in both comparison groups as a result of being related to 2 or more incident cases, the individual is counted in each stratum, except in the age analysis, where an FDR of a younger incident case is given precedence. The age stratification was based on the first quartile of the age at diagnosis distribution of all incident cases for each hematological malignancy. Given the in utero origin of a subset of ALL,62 cases with a high probability of being monozygotic twins were excluded from the analysis.
The lifetime cumulative risk was calculated on the basis of the average life expectancy in Sweden in 2015 (82 years) and the following calculation63 : lifelong cumulative rate = sum of all age-specific incident rates (0-82 years); lifelong cumulative risk = 1–e–Σλi. Exact values for person-years from individual data were used to calculate cumulative incidence.
To estimate the likely contribution of the bone marrow failure and leukemia susceptibility syndromes for which population-based effect sizes exist (ie, Fanconi anemia, Diamond-Blackfan anemia, Schwachman-Diamond syndrome, dyskeratosis congenita, Li Fraumeni, and ataxia telangiectasia) to the FRRs, we made use of published data on their population prevalence and the documented risk for respective hematological malignancies associated with each.64,,,,-69 Using this information and assuming the effect of all mutations being equal, the contribution of each mutated gene to the familial risk for each hematological malignancy was calculated from , where λ0 is the familial risk to first-degree relatives of patients and λk is the familial relative risk associated with the gene mutations k, calculated as , where pk is the risk allele frequency k, qk = 1−pk, and rk is the published relative risk.70
We estimated the power to demonstrate different relative risks as per Newman,71 based on the population incidence rate and number of person-years, stipulating a P value of .05 (2-sided).
Statistical analyses were performed using Stata version 14 (STATA, College State, TX) and R 3.3.3 software. A P value ≤ .05 (2-sided) was considered statistically significant.
Results
Overall estimates
Of the 16.1 million individuals registered in the Swedish Family-Cancer Database, 153 115 individuals were diagnosed with a major classifiable primary hematological malignancy between 1958 and 2015. Supplemental Table 1, available on the Blood Web site, shows the characteristics of cases and the FDRs. Familial cases represented 4.1% of all hematological malignancy diagnoses, which is higher than cancers of the nervous system (1.8%), kidney (2.8%), and pancreas (3.0%) ,but lower than those of the breast (8.5%), colorectum (10.1%), and prostate (15.3%).72
Familial aggregation of hematological malignancies
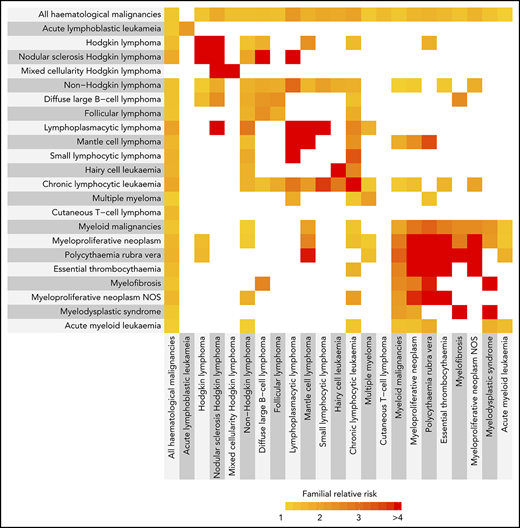
Almost all of the hematological malignancies showed statistically significant increased familial risks for the same tumor type (Figure 1; supplemental Table 2). We have reported a detailed analysis of the FRRs of the myeloid malignancies in this population.18 Briefly, we found 1.5-, 6.8-, 6.9-, and 7.7-fold increases in FRRs for AML, ET, MDS, and PV. For the B-cell tumors, a range of FRRs was observed, with 2-fold increases in FRRs for DLBCL, FL, and MM and 5.6-, 8.3-, 9.8-, 13.3-, 15.8-, and 16.7-fold increases in FRRs for CLL, hairy cell leukemia, nodular sclerosis HL, MCL, LPL/WM, and mixed cellularity HL. We were not able to provide evidence to support familial clustering of CML, myelofibrosis, and the T-cell neoplasms.

The inter-relationship between familial relative risks for different hematological malignancies. The color corresponds to the magnitude of the familial risk as indicated. White indicates a familial risk that crosses unity (nonsignificant). HL comprises nodular sclerosis HL and mixed cellularity HL. NHL comprises DLBCL, FL, LPL, MCL, small lymphocytic lymphoma, hairy cell leukemia, and the T-cell leukemias/lymphomas. Myeloid malignancies comprise PV, ET, myelofibrosis, MPN-NOS, MDS, CML, and AML. Myeloproliferative neoplasms comprise PV, ET, myelofibrosis, MPN-NOS. Hematological malignancies with no significant familial associations are not shown.
The inter-relationship between familial relative risks for different hematological malignancies. The color corresponds to the magnitude of the familial risk as indicated. White indicates a familial risk that crosses unity (nonsignificant). HL comprises nodular sclerosis HL and mixed cellularity HL. NHL comprises DLBCL, FL, LPL, MCL, small lymphocytic lymphoma, hairy cell leukemia, and the T-cell leukemias/lymphomas. Myeloid malignancies comprise PV, ET, myelofibrosis, MPN-NOS, MDS, CML, and AML. Myeloproliferative neoplasms comprise PV, ET, myelofibrosis, MPN-NOS. Hematological malignancies with no significant familial associations are not shown.
We next examined FRRs of the lymphoid diseases by age at diagnosis, sex, and type of familial relationship. Familial risks were significantly higher for relatives of cases diagnosed young for all HL (5.76 vs 3.36) and CLL (6.99 vs 4.83; Table 1). The FRRs were significantly higher in siblings when compared with that of parent-offspring relationships for NHL (1.97 vs 1.69), HL (7.45 vs 3.09), and CLL (7.80 vs 5.36). In contrast, parent-offspring RRs were significantly higher for LPL/WM (21.88 vs 5.56; supplemental Table 3). We found no evidence for differences in FRR by sex (supplemental Tables 4 and 5). FRRs were significantly higher for relatives with 2 or more affected FDRs when compared with relatives with 1 affected FDR for all hematological malignancies (2.08 vs 1.31) and CLL (27.13 vs 5.36).
As shown for the myeloid malignancies,18 familial risks were significantly higher for relatives of cases diagnosed young for all MPNs (6.46 vs 4.15), PV (10.91 vs 5.96), and MDS (11.95 vs 3.27; Table 1). Sibling relative risks were higher than parent-child relative risks for AML (3.08 vs 1.09), and all 53 familial cases of PV were of a parent-child relationship. Familial relative risks were significantly higher for relatives with 2 or more affected FDRs for all myeloid malignancies (4.55 vs 1.96) and all MPNs (17.82, 4.83).
Cumulative risk
Supplemental Table 6 details the cumulative risk for each of the hematological malignancies. The lifetime cumulative risks for all primary hematological malignancies, NHL, and CLL in the Swedish population were 3.4%, 1.3%, and 0.4%, respectively (Table 2; supplemental Table 6). As the FRRs estimates for the hematological malignancies are relatively high compared with the FRRs of other cancers,73 the lifetime cumulative risk in FDRs of patients increased to 4.3%, 2.2%, and 2.3% for all primary hematological malignancies, NHL, and CLL, respectively (Figure 2). For individuals with 2 or more affected FDRs, markedly elevated cumulative risk estimates for all primary hematological malignancies (7.4%) and CLL (8.6%) were observed (Table 2).
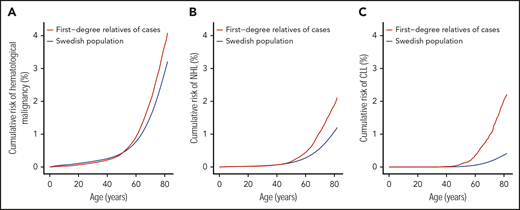
Lifetime cumulative risk for all hematological malignancies, NHL, and CLL. (A) All hematological malignancies, (B) NHL, and (C) CLL.
Lifetime cumulative risk for all hematological malignancies, NHL, and CLL. (A) All hematological malignancies, (B) NHL, and (C) CLL.
Contribution of cancer syndromes to familial risk
A number of rare bone marrow failure and leukemia susceptibility syndromes caused by inheritance of germline mutations influence the risk for specific hematological malignancies.19 For example, ataxia telangiectasia is associated with an increased risk for ALL and AML.69 Others that are rarer include Fanconi anemia,64 Diamond-Blackfan syndrome,65 and dyskeratosis congenita.66 Thus far, no high-impact mutations have consistently been shown to influence the risk for the mature B-cell neoplasms in adults. The rare bone marrow failure and leukemia susceptibility syndromes contribute 17%, 7%, and 4% of the population-based familial risk estimates for MDS, ALL, and AML, respectively (supplemental Figure 1).
Shared familial risk across hematological malignancies
To explore the possibility of familial aggregation between different tumor types, we examined FRRs between all hematological malignancies. Although the strongest FRRs tended to be disease-specific, we also identified distinct patterns of familial risk (Figure 1; supplemental Table 2). These were predominantly cell-lineage specific; for example, CLL shared familial risks with nearly all the B-cell tumors. However, familial risks were also noted across cell lineage, with HL, DLBCL, MCL, CLL, and MM sharing risks with a number of myeloid malignancies. In contrast, no familial aggregation was observed for CML, Burkitt lymphoma, anaplastic T-cell lymphoma, and mature T-cell lymphoma (Figure 1; supplemental Table 2).
Discussion
This study provides further support for the significant familial aggregation of the major hematological malignancies. Because of the large sample size and long follow-up time, we have been able not only to demonstrate significantly elevated relative risks in FDRs of cases for the same tumor type but also to detect associations between the different hematological tumor types. Although we were able to capture the major histological subtypes of the hematological malignancy as defined by ICD-7 and ICD-O/2 codes, the study of FRRs using more recent developments to the classification system may further refine FRR estimates.1,2
Our analysis benefits greatly from being based on the Swedish Family-Cancer Database. This population-based family cancer registry possesses near-complete case registration for almost all hematological malignancies, thereby allowing familial relative risks to be derived while avoiding biases introduced by case-control study designs.74,75 Estimates of underreporting of cancer diagnoses are thought to be stable over periods and have been estimated as 4% in 1978, 3.7% in 1998, and 4% in 2014.74,76,77 Furthermore, our analysis is enhanced by the large number of cases when compared with earlier studies, which have previously exploited this resource, particularly in respect to the hematological diseases that have been classified from 1993.52,54,78 Although the strengths of our study are the large sample size and unbiased assessment of cancer status in relatives, we did not have information concerning other possible risk factors for malignancies that may be correlated within families.
The familial risk estimates provided in this study are broadly comparable to those previously published from the Swedish population.15,-17,51,,,-55 One notable difference is the discrepancy in familial risk for DLBCL.50 This difference may be a result of previous analyses based on a case-control design with a consequential inflation of risk estimates, definition of affected relatives, and greater case numbers with longer follow-up in this study.
Although elevated familial risks cluster with diseases from the same hematopoietic cell lineage, a number of associations extend across hematopoietic cell lineages. These findings suggest that etiological factors for hematological malignancies are shared not only between diseases arising from cells at different stages of differentiation of the same hematopoietic lineage but also between diseases originating from different hematopoietic lineages. It is likely that there is heterogeneity in the mechanisms by which these factors exert their effects on different phenotypes, as evidenced by the observation that the majority of FRRs are highest for concordant disease, and that some of the risks differed depending on the relationship of the FDR.
The association between familial risk and age of onset for many of the hematological malignancies provides support for the role of genetic predisposition influencing disease risk.49 This was especially marked for PV, MDS, HL, and CLL, which displayed high FRRs for relatives of cases and for those related to patients diagnosed young. The genetic architecture of susceptibility to hematological malignancy is not fully defined. Rare high-impact mutations in a number of genes have been identified, conferring elevated risks for myeloid and/or lymphoproliferative malignancies.19,-21,23,-25,28,31,-33,79,,,,,,,,-88 Our analysis indicates that the known genetic predisposition syndromes for which population-based effect estimates exist are unlikely to account for the observed FRRs. Although the existence of additional major genes, as well as more recently described mutations, may account for part of the observed FRR, it is likely that some of the FRR is enshrined in common genetic variation.49 Indeed, although few published studies have investigated the role of such variation in MDS and AML, common genetic variants have already been shown to contribute significantly to the FRR of the other hematological malignancies.36,43,44,89,90 Albeit preliminary, evidence for a shared susceptibility between different hematological malignancies consistent with our observations has been proposed from recent analyses of GWAS data.91,92 A component of the FRR may also be explained by shared environmental risk factors, although the magnitude of their role in explaining familial aggregation has been debated.93,,-96 To date, few robustly identified environmental risk factors for hematological malignancy have been described, with examples including infection with EBV, HIV, HTLV, and Helicobacter pylori, as wel as benzene, cytotoxic therapy, and ionizing radiation.9,,-12 Of note, there are some, albeit nonconclusive, data, to suggest ionizing radiation may increase the risk for lymphoid as well as myeloid malignancies.11,97,-99
Although the FRRs associated with some of these hematological malignancies are high compared with those associated with the solid cancers,73 the absolute risk is small (Table 2). For example, given that the population lifetime risk for CLL is only 0.4%, an FRR of 6.99 translates to a risk of 2.2% for a first-degree relative of a CLL case. Such familial risks may therefore be of limited clinical significance, with a few notable caveats. First, a diagnosis of CLL represents a proportion of the observable CLL-related phenotype. Monoclonal B-lymphocytosis, the precursor of CLL, is present in 3% to 4% of adults older than 40 years and 14% to 18% of FDRs of CLL.100 An increase in prevalence of such precursor diseases in relatives is therefore likely to be observed for myeloma and the myeloid malignancies with respect to the precursor lesion monoclonal gammopathy of unknown significance and clonal hematopoiesis, respectively.101,,,,,-107 Second, FRRs reflect the influence of all forms of family history on risk. Inevitably, this may include a subset with clear Mendelian inheritance. This is reflected in the greater lifetime risk associated with having multiple affected relatives of more than 4% and 8% for the myeloid malignancies and CLL, respectively, and is consistent with the existence of genes with stronger effects such as those predisposing to AML, MDS, and CLL.19,79 The significant familial aggregation shown here therefore justifies the continued application of gene-mapping approaches in high-risk families and suggests that within families, this may lead to identification of additional high-penetrance mutations.
There are a number of limitations to our analysis. First, our findings may only apply to Western countries and may not therefore be applicable to economically developing countries that have different tumor incidence rates.108,,-111 Second, although we had good power to demonstrate modest FRRs (ie, greater than 80% power for an FRR of 1.5) associated with the major classes of hematological malignancies (ALL, CLL, AML, CML), for the malignancies only cataloged post-1993, we have had limited power (eg, only 50% power for an FRS of 2.5 in the case of LPL/WM; supplemental Figure 2). Finally, although we have sought to reduce the effect of misclassification of cancer outcomes by applying ICD-O/2 and SNOMED codes and the use of synonymous classification terms across time, there remains a potential for misclassification to affect estimates.
Accepting these caveats, the results of our analyses have important implications. First, these results can inform initiatives to identify and characterize etiological risk factors for hematological malignancy. Such efforts offer the prospect of providing insight into disease biology.49,112,,,-116 Furthermore, given the presence of disease nonconcordant FRR, it may be appropriate, with careful study design, to expand such initiatives to include multiple hematological phenotypes. Second, given that even moderate familial associations indicate large differences in risk between individuals in the population,47,117 such risks can improve the management of patients with hematological malignancy and their relatives through counseling and genetic testing.19,118
In summary, we have performed a comprehensive analysis of familial risks of the major hematological malignancies in the Swedish population. These results have the potential to assist in the management of patients with hematological malignancy and their relatives and to inform future studies investigating the etiological basis of these hematological malignancies.
These data are not publicly available because of restrictions (information that could compromise participant privacy).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This project was supported by grants from the German Cancer Aid, the Swedish Research Council (2014-2517, 2014-10134 and 2016-01176), ALF funding from Region Skåne. A.S. is the recipient of a guest scientist Fellowship from DKFZ. The work of R.S.H. is supported by funding from Bloodwise.
Authorship
Contribution: A.S., R.S.H., and K.H. designed the study; K.S., J.S., and K.H. provided the data; A.S., S.C., and H.T. performed data extraction and statistical analysis; A.S., R.S.H., and K.H. drafted the manuscript; and all authors contributed to the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Amit Sud, Division of Genetics and Epidemiology, The Institute of Cancer Research, 15 Cotswold Rd, London SM2 5NG, United Kingdom; e-mail: amit.sud@icr.ac.uk.