TO THE EDITOR:
Philadelphia (Ph)-like acute lymphoblastic leukemia (ALL) is a high-risk subtype of B-precursor ALL (B-ALL) characterized by a gene-expression profile similar to Ph+ ALL but lacking the specific BCR-ABL1 fusion gene.1,2 Patients with Ph-like ALL are at higher risk of induction failure or high postinduction minimal residual disease (MRD) levels.3-5 This poor early response translates into inferior outcome in event-free survival (EFS) and overall survival (OS) as compared with other B-ALL patients. Ph-like ALL was recognized as a provisional entity by the 2016 World Health Organization (WHO) classification but strict and unequivocal diagnosis criteria have not yet been established.6 Ph-like ALL harbors diverse oncogenic lesions that all lead to the aberrant activation of cytokine receptors or signaling factors, the most frequent being rearrangements of CRLF2, fusions and mutations of JAK kinases, and fusions involving ABL-class kinases (ABL1, ABL2, CSF1R, or PDGFRB).7 As anticipated, Ph-like cells harboring ABL-class rearrangements have shown sensitivity to tyrosine kinase inhibitors (TKIs) such as imatinib and dasatinib in vitro or in patient-derived xenograft.8-10 In addition, we and others have reported single experiences of clinical benefit of TKI treatment in early resistant patients.11,12
In an attempt to enable targeted therapy in B-ALL patients with poor response to chemotherapy, we developed an integrative diagnosis strategy to identify Ph-like alterations in newly diagnosed or relapsing patients in a timely manner. We report on 24 patients with B-ALL harboring ABL-class fusions, initially enrolled in or treated according to pediatric (French Acute Lymphoblastic Leukaemia Study Group [FRALLE]/French Protocol for the Treatment of Acute Lymphoblastic Leukemia in Children and Adolescents [CAALL]/European Organisation for Research and Treatment of Cancer [EORTC]) (NCT02716233; NCT01185886) or adult (French-Swiss-Belgian Group for Research on Adult Acute Lymphoblastic Leukemia [GRAALL]/European Working Group on Adult ALL [EWALL]) (NCT00327678; NCT02617004) trials, who could be treated with a combination of TKI and chemotherapy, either during frontline treatment (N = 19) or at relapse (N = 5). Cytogenetic analyses were performed locally. Molecular analyses were performed centrally using in-house, multiplexed targeted methods13 for detection of fusion transcripts, and in a second step, RNA sequencing for unresolved cases. The current algorithm of our screening strategy is shown in supplemental Figure 1 (available on the Blood Web site).
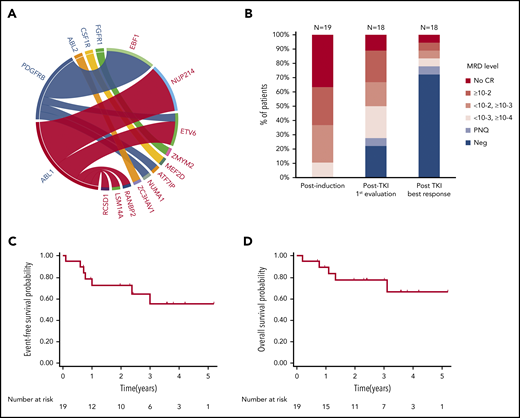
Of the 24 patients reported here, 12 had fusions involving ABL1, including NUP214-ABL1 (n = 6), ETV6-ABL1 (n = 3), and other partners in single cases (RCSD1, RANBP2, and LSM14A) (Figure 1A). Nine cases had a PDGFRB rearrangement, including EBF1-PDGFRB (n = 6) and other partners in single cases (NUMA1, ETV6, and ATF7IP). ZC3HAV1-ABL2 and MEF2D-CSF1R fusions were identified in single cases, and a patient with a ZMYM2-FGFR1 fusion (not strictly ABL class) was also included in the present cohort.

Patient characteristics and outcome. (A) ABL-class fusion gene in the whole cohort (N = 24). On the circos plot, the names of ABL-class genes are in blue; partner genes are in red. (B) MRD response in patients treated frontline at different time points (N = 19, 1 patient not evaluated after TKI exposure). EFS (C) and OS (D) from diagnosis in patients exposed frontline to TKI (N = 19).
Patient characteristics and outcome. (A) ABL-class fusion gene in the whole cohort (N = 24). On the circos plot, the names of ABL-class genes are in blue; partner genes are in red. (B) MRD response in patients treated frontline at different time points (N = 19, 1 patient not evaluated after TKI exposure). EFS (C) and OS (D) from diagnosis in patients exposed frontline to TKI (N = 19).
The resulting cohort (Table 1) included 16 male and 8 female patients; the median age at diagnosis was 24 years (range, 5-72 years). Two patients (#1 and #24) were previously reported.11,14 As expected, patients had baseline characteristics and early response to treatment associated with a poor prognosis. The median white blood cell count was 30 × 109/L (range, 4 × 109/L to 570 × 109/L). Intragenic IKZF1 deletions were detected in 11 of 24 patients (46%). A poor response to prednisone prephase (≥1 × 109/L blasts in peripheral blood at day 8) was observed in 14 of 22 evaluable patients (64%). After induction therapy, only 16 of 24 patients (67%) reached complete remission (CR), all with detectable MRD, including 7 with MRD ≥10−2 (Table 1; Figure 1B).
In 19 patients, the ABL-class fusion was identified at initial diagnosis workup and TKI was introduced during frontline treatment, most frequently within the first month of consolidation or salvage (median 49 days from diagnosis). In 5 patients, ABL-class fusions were diagnosed at relapse and TKI was introduced in association with salvage therapy. Remarkably, 2 of these patients had postinduction MRD below 10−4 during frontline treatment. In this study, the physician chose the TKI, TKI dosage, and combination. Fourteen patients were exposed to imatinib, 9 to dasatinib, and 1 to ponatinib. Three patients were switched from imatinib to dasatinib during frontline treatment (Table 1).
Among the 19 patients treated with TKI frontline, 6 of 7 primary refractory patients subsequently achieved CR, 1 after a switch to dasatinib (#10). One patient died early of sepsis in a context of uncontrolled disease (#16). One patient was lost of follow-up after salvage until she relapsed (#13). In 14 of 18 patients (78%), an MRD level below 10−4 was achieved within a median time of 2.5 months (range, 1.4-14.8 months) after TKI initiation (Figure 1B). Allogeneic hematopoietic stem cell transplant (HSCT) was performed in 9 patients, 3 with a sibling donor, 4 with a matched unrelated donor (MUD), and 2 with a haploidentical donor (Haplo). All patients were in CR before HSCT and 6 of 9 (67%) had undetectable MRD. One patient was additionally exposed to blinatumomab in combination to dasatinib in bridge to HSCT. After a median follow-up of 36 months (range, 8-73 months), 12 patients were alive in first CR. Six patients relapsed; 3 patients received an alternative TKI, including 1 in association to inotuzumab. The median remission duration and OS were not reached. At 3 years, EFS was 55% (95% confidence interval, 27-76) and OS was 77% (95% confidence interval, 50-91) (Figure 1C-D).
The 5 patients who were treated with TKI at relapse achieved CR, including 2 patients who had refractory disease to several lines of treatment (#22 and #24). An MRD level below 10−4 was achieved in 3 patients, of whom 2 could proceed to HSCT (1 haploidentical donor, 1 matched unrelated donor) and remained alive in remission. The 3 other patients further relapsed and died of progressive disease after exposure to second-line TKIs (dasatinib, n = 2; ponatinib, n = 1).
Although anecdotal reports of clinical efficacy of TKIs in patients with ABL-class mutant Ph-like ALL have been described,8,11,12,15-17 more systematic TKI introduction has been severely hampered by the diagnostic challenge of detecting previously unknown 5′ fusion partners in a timely manner. We report here a significant cohort of patients for whom identification of ABL-class fusions led to early introduction of TKI therapy. Previous adult studies have reported the poor outcome of Ph-like ALL in adults with 5-year OS ranging from 22% to 27%.3-5 The 3-year OS of 77% we observed in the patients treated frontline compares favorably with these retrospective observations. Beyond the use of historical cohort, this comparison has, however, some limitations. Indeed, the historical series included all Ph-like ALL cases characterized by gene-expression profile, and no specific subgroup analysis was performed in patients with ABL-class fusion genes. The outcome of non-CRLF2-rearranged patients was suggested to be better than CRLF2-rearranged cases, which may lead to underestimation of the outcome of ABL-class mutant Ph-like ALL treated without TKI.4 However, considering the highly resistant profile of our patients after induction, the outcome with conventional therapy was expected to be poor. Finally, the outcome of children and adolescents, a minority of patients in our cohort, has been reported to be better than in adults, which may reflect the use of more intensive chemotherapy regimen.8
In conclusion, we report the largest cohort of patients with ABL-class kinase rearrangement exposed to TKI frontline or at relapse, and show promising MRD response and outcome, reminiscent of those observed in early trials of imatinib combined with chemotherapy in Ph+ ALL.18 Prospective screening strategies are feasible and should be generalized to identify these high-risk patients and to propose early TKI-based intervention. In future studies, several questions remain to be addressed, including the choice of TKI according to the fusion transcript, whether these patients should receive recently approved blinatumomab,19 and finally the benefit of HSCT in patients who achieve good MRD response upon targeted therapy.
The online version of this article contains a data supplement.
Acknowledgment
This work was supported by a grant from Le Fonds de Dotation Contre la Leucémie (JNCL 2014-01).
Authorship
Contribution: I.T. collected and analyzed data and wrote the manuscript; I.B. performed experiments and analyzed data; N.S., T.B., A.T.-S., J.T., S.M., E.D., C.H., C.D., P.C., P.R., A.B., J.L.-P., and N.B. treated patients and provided clinical data; W.C., P.B., N.D., and H.C. provided molecular and cytogenetic data; M.B. analyzed data and wrote the manuscript; H.D. and J.S. contributed to the study design and wrote the manuscript; N.B. and E.C. designed research, collected and analyzed data, and wrote the manuscript; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Nicolas Boissel, Hematology Department, Saint-Louis Hospital, 1 Ave Claude Vellefaux, 75010 Paris, France; e-mail: nicolas.boissel@aphp.fr.