Key Points
Secondary ITP complicates the clinical course of chronic LPDs in up to 5% of patients and is poorly responsive to conventional treatments.
Eltrombopag is effective in increasing platelet count to safe levels in most patients, thus postponing otherwise unnecessary chemotherapy.

Abstract
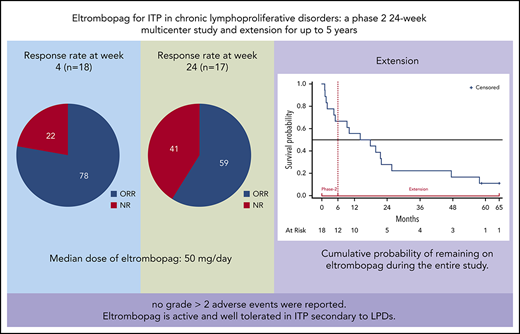
Immune thrombocytopenia (ITP) secondary to chronic lymphoproliferative disorders (LPDs) is poorly responsive to conventional treatments. We conducted a multicenter phase 2 prospective 24-week study in 18 patients with ITP secondary to LPDs to assess the safety and efficacy of eltrombopag. Responsive patients entered an extension study for up to 5 years. For inclusion, patients should not require cytotoxic treatment and should have a platelet count <30 × 109/L or have symptoms of bleeding. Eltrombopag was initiated at 50 mg/day, with a maximum of 150 mg/day. The primary end point was platelet response after 4 weeks. Median age was 70 years (range, 43-83 years), and 14 patients had chronic lymphocytic leukemia, 2 had classic Hodgkin lymphoma, and 2 had Waldenström macroglobulinemia. All patients had received previous ITP treatments. Response rate at week 4 was 78% (95% confidence interval [CI], 58%-97%), with 50% of patients having a complete response (CR) (95% CI, 43%-57%); respective results at week 24 were 59% (95% CI, 36%-82%) with 30% reaching a CR (95% CI, 8%-52%). Median exposure to eltrombopag was 16 months; median dose at week 4 was 50 mg/day (range, 25-100 mg/day), and at week 24, it was 50 mg/day (range, 25-150 mg/day). No grade >2 adverse events were reported. Eltrombopag is active and well tolerated in ITP secondary to LPDs. This trial was registered at www.clinicaltrials.gov as #NCT01610180.
Introduction
Eltrombopag (Revolade), an orally bioavailable thrombopoietin receptor-agonist (TPO-RA), is a consolidated treatment for primary immune thrombocytopenia (pITP),1-3 but it has been poorly investigated in secondary ITP (sITP) of chronic lymphoproliferative disorders (LPDs). sITP is the second most frequent autoimmune cytopenia in LPDs after hemolytic anemia.4-7 Thrombocytopenia in LPDs may confound staging, increase bleeding risk, or prompt an otherwise unnecessary treatment of the underlying disease. Unfortunately, sITP of LPDs has proved to be more resistant to conventional treatments than pITP.5,8-11 On the premise of a common pathogenic autoimmune mechanism, we investigated the efficacy and safety of eltrombopag for increasing platelet count in patients with sITP of LPDs.
Patients and methods
This was a phase 2, open-label, 24-week long, single-arm multicenter prospective study. The primary end point was platelet response (R) at week 4. At the physician’s discretion, responding patients entered an extension phase for up to 5 years. Eighteen patients were enrolled in 7 centers in Italy between September 2012 and November 2015 and were observed until June 2018 or before if they met the discontinuation criteria. The institutional review boards approved the study, and patients gave informed consent. Patients with chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma, or other B-cell lymphoproliferative disorders according to World Health Organization 2008 criteria12 and sITP according to predefined criteria were eligible (supplemental Table 1 available on the Blood Web site).
A Simon’s optimal 2-stage design was adopted.13 Under a null hypothesis H0: p0 ≤ 0.20 and an alternative hypothesis H1: p1 ≥ 0.50, continuing enrollment was allowed on the basis of the responses in the first 8 patients; 18 patients were required for a one-sided 5% α error and a 20% β error. One unresponsive patient, who later became ineligible, was conservatively maintained for outcome calculations.
Eltrombopag treatment was initiated at 50 mg/day for 14 days. Subsequent dose adjustments of ±25 mg were made once per week from weeks 3 to 6, and subsequently every 2 to 4 weeks if the dose was stable during the preceding 4 or 8 weeks. The maximum dose was 150 mg/day. Eltrombopag was kindly provided by GlaxoSmithKline (Verona, Italy) until March 2015; then it was provided by Novartis (Origgio, Italy). Additional inclusion or exclusion criteria, amendments, study requirements, visit frequency, clinical investigations, response, and discontinuation criteria during the study and statistical plan are fully reported in the supplemental Data.
Results and discussion
Patient’s characteristics at study entry are provided in supplemental Table 2. All patients had been treated with corticosteroids for thrombocytopenia before enrollment, and all showed insufficient response. Treatment with corticosteroids, if it was ongoing at enrollment, should have been at a stable dose for at least the preceding 3 weeks, and patients should have stopped receiving lymphoma treatment for at least 1 month. During phase 2, the primary end point was achieved in 14 patients (78%) who reached R or complete response (CR) at week 4, which was maintained at week 24 in 59%. CRs were 50% and 30%, at weeks 4 and 24, respectively, as detailed in Table 1. Fifteen patients achieved sustained continuous response for at least 4 weeks at a dose range of 12.5 to 150 mg/day, and 3 patients achieved sustained continuous response while receiving 12.5 to 25 mg/day. The median dose was 50 mg/day (range, 12.5-150 mg/day) (supplemental Figure 1). Median duration of continuous R was 18 weeks (interquartile range, 11-22 weeks); the duration for CR was 15 weeks (interquartile range, 4-18 weeks). Distribution of patients by continuous weeks of R or CR is shown in supplemental Figure 2. Response status, safety, and eltrombopag discontinuation during the entire study are shown in Figure 1 for each patient. One nonresponsive patient who did not meet the inclusion criteria, was withdrawn from the study at week 7 (patient 1); 15 patients discontinued treatment: 1 for inefficacy and protocol violation, 8 for loss of response, 3 for death, and 3 for LPD progression. Two were continuing treatment with eltrombopag at the end of the study. The cumulative probability of continuing to receive eltrombopag (median, 16 months; range, 1-58 months) is shown in supplemental Figure 3.
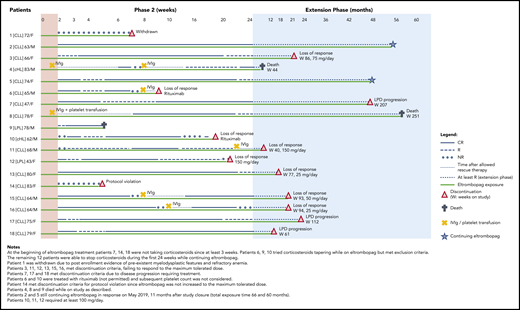
Response status (CR or R for phase 2 and at least R for the extension phase), rescue treatments, exposure to eltrombopag, and discontinuation or death during phase 2 and extension phase for all 18 patients.
Response status (CR or R for phase 2 and at least R for the extension phase), rescue treatments, exposure to eltrombopag, and discontinuation or death during phase 2 and extension phase for all 18 patients.
No grade >2 adverse events, including bleeding, occurred. One patient developed grade 2 unprovoked iliac-femoral venous thrombosis during R at week 12 of treatment with eltrombopag, which was not discontinued. Another patient developed grade 2 itching that was controlled by topical corticosteroids. No grade >1 increment of bone marrow reticulin or transaminitis occurred.
After an amendment excluded patients with >3% CD34+ immature cells on bone marrow biopsy, no increase of CD34+ cells or myelodysplastic features were observed. Three deaths were recorded, all in responsive patients. A 78-year-old man with a 5-year history of LPL (patient 9) died while in CR at week 5 of therapy with eltrombopag at 75 mg/day as a result of complicated diverticulitis and intestinal perforation complicated by septic shock. Autopsy revealed transformation to aggressive B-cell lymphoma involving bone marrow and the gastrointestinal tract. A heavily pretreated 83-year-old man with classic Hodgkin lymphoma and several cardiac comorbidities (patient 4) died of pneumonia during the third month of the extension while receiving eltrombopag at 50 mg/day. A 78-year-old female with CLL (patient 8) died as a result of disease progression and multiorgan failure on the 58th month of extension while receiving eltrombopag at 50 mg/day. Three patients (7, 17, and 18) required treatment for progression of their CLL while they were still responding to eltrombopag. All events were considered unrelated to the study drug.
Efficacy of eltrombopag in sITP was comparable to that reported in pITP2,14 with a median dose of 50 mg/day. None of the responsive patients required doses above 100 mg/day. Increasing the dose to 150 mg/day, as is currently approved in severe aplastic anemia,15 was ineffective in 2 patients who were unresponsive to 100 mg/day. Recently, a phase 2 trial of eltrombopag for patients with CLL and thrombocytopenia (that used response criteria adopted for myelodysplasia) showed an overall response similar to that in our study in the 11 patients with CLL-associated ITP who were observed for a shorter time (range, 1.8-9.4 months).16 The maximum dose of eltrombopag was higher (300 mg/day), with possibly treatment-related transaminitis in 2 patients; a case of lower limb deep vein thrombosis complicated by pulmonary embolism was also reported. Successful use of TPO-RA in patients with CLL-associated ITP refractory to standard treatment for ITP have been also described by only a few reports for which publication bias cannot be excluded.14,17-20 TPO-RA might potentially stimulate neoplastic cells, as reported in patients with myelodysplasia or promote clonal expansion as in aplastic anemia.21 Thus, we conservatively considered ≤3% CD34+ count in the bone marrow a mandatory prerequisite for patients who were candidates for treatment with TPO-RA in the setting of sITP of LPDs. Overall, no evident adverse effects were noted during the clinical course of the underlying LPD (Figure 1). Patient 9 developed histologic transformation of LPL in large B-cell lymphoma. Although the contribution of eltrombopag cannot be excluded, transformation of LPL to high-grade lymphoma is a well-documented complication, with median time to transformation of 4.6 years and frequent involvement of extranodal tissues,22 as in our patient.
Our prospective study, although limited by the lack of a comparative arm and the relatively small number of patients, most of whom were affected by CLL is, to our knowledge, the only study that estimated the efficacy of eltrombopag according to widely accepted standardized criteria used in ITP23 and that observed patients for a long enough time to estimate the impact of eltrombopag on the overall clinical course of LPDs. Further prospective studies comparing eltrombopag to standard of care are needed to confirm our findings on the efficacy of this treatment and also to expand our knowledge on its safety, including the potential increased risk of thrombosis, which has been reported to be likely associated with ITP and the use of TPO-RA.24
For original data, please contact the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgment
The authors thank Lisanna Ghiotto and Andrea Timillero for their help in analyzing the data and drawing the figures and Claudia Guzzoni for her invaluable secretarial assistance (all from the Hematology Project Foundation).
This study was funded by the Hematology Project Foundation (Vicenza, Italy).
Authorship
Contribution: C.V. and F.R. contributed equally to conceiving and designing the study, collecting and interpreting the data, and writing the protocol and manuscript; A.B., G.C., E.S.G.d'A., E.L., G.M., M.M., F.P., G.Q., A. Romano, A. Rambaldi, M.R., R.M.S., F.V., and S.V. collected data; and all authors had access to primary clinical data and reviewed and approved the manuscript.
Conflict-of-interest disclosure: F.R. served on the speaker’s bureau for Novartis and Amgen and serves as a consultant for Argenx. F.P. served on the advisory board and speaker’s bureau for Novartis. A.B. served on the speaker’s bureau for Novartis and Amgen. The remaining authors declare no competing financial interests.
Correspondence: Francesco Rodeghiero, Hematology Project Foundation, Contrà San Francesco 41, 36100 Vicenza, Italy; e-mail: francesco.rodeghiero@hemato.ven.it.