Key Points
There was no difference in outcomes after MMRD and MUD HSCT with TCRαβ+/CD19+ graft depletion in patients with primary immunodeficiencies.
HSCT from MMRDs with TCRαβ++/CD19+ graft depletion is a safe and effective alternative for MUD HSCT in patients with PID.

Abstract
TCRαβ+/CD19+ graft depletion effectively prevents graft-versus-host disease (GVHD). In the current study, we compared the outcomes of hematopoietic stem cell transplantation (HSCT) with TCRαβ+/CD19+ depletion from matched unrelated donors (MUDs) and mismatched related donors (MMRDs) in patients with primary immunodeficiency (PID). A total of 98 pediatric patients with various PIDs underwent HSCT with TCRαβ+/CD19+ graft depletion from MUDs (n = 75) and MMRDs (n = 23). All patients received a fludarabine-/treosulfan-based conditioning regimen, with 73 also receiving a second alkylating agent. For GVHD prophylaxis, all but 2 received serotherapy (antithymocyte globulin) before HSCT and a short course of posttransplant immunosuppression. Neutrophil and platelet engraftment in both the MUD and MMRD groups occurred on days 14 and 13, respectively. The incidence of secondary graft failure was 0.16 and 0.17 (P = .85), respectively. The cumulative incidence of acute GVHD grade 2 to 4 was 0.17 in the MUD group and 0.22 in the MMRD group (P = .7). The incidence of cytomegalovirus (CMV) viremia was 0.5 in the MUD group and 0.6 in the MMRD group (P = .35). The frequency of CMV disease was high (17%), and the most common manifestation was retinitis. The kinetics of immune recovery was similar in both groups. The overall survival was 0.86 in the MUD group and 0.87 in the MMRD group (P = .95). In our experience, there was no difference in the outcomes of HSCT performed from MUD and MMRD. Hence, given the immediate availability of donors, in the absence of HLA-identical siblings, HSCT with TCRαβ+/CD19+ graft depletion from MMRDs can be considered as the first choice in patients with PID.
Introduction
Primary immunodeficiencies (PIDs) are rare disorders caused by mutations of various genes essential for the development and function of immune system components. PIDs phenotypically present with infections, autoimmunity, and malignancies.1 For many PIDs, the only curative method is hematopoietic stem cell transplantation (HSCT). During the last decade, the list of indications for HSCT has rapidly grown to include a significant number of PID types. For many years, the choice of donor for HSCT in patients with PID without HLA-identical siblings has remained a serious problem. The use of alternative donors has been associated with higher risks for posttransplant complications and increased morbidity and mortality.2 Severe graft-versus-host disease (GVHD) is 1 of the major obstacles in HSCT from matched unrelated donors (MUDs).3 The use of different methods of T-cell elimination from the graft in haploidentical transplantation allows for a decreased incidence of GVHD,4 but at the same time, profound depletion of immunocompetent cells results in delayed immune recovery, which correspondingly increases the risk for severe infectious complications and significantly decreases survival as a result. The issue of delayed immune reconstitution is especially critical for patients with PID, who are typically infected with multiple pathogens before transplantation.2 TCRαβ+/CD19+ depletion is a relatively new precision technology of graft engineering that, in a few studies conducted, demonstrated effective prevention of GVHD in patients with PID with a high survival rate after both haploidentical and MUD HSCT.5-8
Here we present a comparison of single-center results of HSCT with TCRαβ+/CD19+ graft depletion from MUDs and mismatched related donors (MMRDs) in a representative cohort of pediatric patients with PID.
Methods
This trial was registered at www.clinicaltrials.gov as a prospective, interventional, single-group, open-label study (NCT02327351). The research was approved by the institutional ethics committee, and patients or their legal guardians signed informed consent forms to participate. Data on 42 patients have been previously published.6,8-11
Subjects included 98 patients who had various PIDs but no nonaffected HLA-identical sibling donors. In 59 of these cases, molecular genetic testing identified the causative mutation. All patients underwent their first HSCT from either MUDs or MMRDs at the Dmitry Rogachev National Medical Center of Pediatric Hematology, Oncology and Immunology between June 2012 and December 2017. Grafts from both MUDs and MMRDs were depleted of TCRαβ+/CD19+ to prevent GVHD. Patients with severe combined immunodeficiency and Nijmegen breakage syndrome were excluded from the analysis because of different risks for mortality associated with multiple uncontrolled infections, including bacilli Calmette Guerin (BCG) in patients with severe combined immunodeficiency and different conditioning regimens in Nijmegen breakage syndrome. Second transplantation procedures after graft failure were performed with different conditioning regimens and GVHD prophylaxis methods, and were not included in this study.
Fifteen patients, 11 in the MUD group and 4 in the MMRD group, had uncontrolled active infectious or autoimmune complications with organ dysfunctions at the time of transplantation. These did not include uncomplicated immune cytopenias, as well as thrombocytopenia in patients with Wiskott-Aldrich syndrome (WAS).
HSCT was performed on 75 patients with 8-10/10 HLA-matched MUDs and on 23 patients with MMRDs. The median follow-up (FU) after HSCT in survivors was 2.5 years (range, 0.5-5.8 years). The main characteristics of the MUD and MMRD groups are presented in Table 1.
HLA tissue typing of patients and MMRD until 2017 was performed by sequencing-based typing (AlleleSEQR, GenDX) and from 2017 on by next-generation sequencing (MiSeq platform, reagents NGSgo, GenDX).
Graft processing
Hematopoietic stem cell grafts were derived from the granulocyte colony-stimulating factor (G-CSF)-mobilized peripheral blood mononuclear cell fraction. Depletion of TCRαβ+ T cells and B cells was performed with CliniMACS Plus and CliniMACS Prodigy instruments (Miltenyi Biotec, Bergisch Gladbach, Germany). Graft composition did not vary significantly in the MUD and MMRD groups, and is presented in Table 1.
Conditioning regimens and posttransplant GVHD prophylaxis
All patients received treosulfan-based conditioning regimens in combination with fludarabine. Because of a high incidence of graft rejection6 after treosulfan/fludarabine-containing regimens, a second alkylator was added to conditioning; in 52 patients it was melphalan or thiotepa, and in 4 it was cyclophosphamide. Seventeen patients with WAS resulting from high incidence of graft rejection also received, in preparative regimens, a combination of G-CSF with plerixafor.8 Detailed information about the conditioning regimens is represented in Table 2.
Ninety-six patients received pretransplant serotherapy. In 2 patients with severe cytomegalovirus (CMV) infections at HSCT time, no serotherapy was used, and the second alkylating agent was switched to cyclophosphamide. Rituximab was used in 76 patients to prevent Epstein-Barr virus (EBV)-associated posttransplant lymphoproliferative disease (Table 2). All patients received short-term posttransplant GVHD prophylaxis until days +45 to +60 (supplemental Table 1, available on the Blood Web site).
Engraftment and graft failure assessment
Neutrophil engraftment was registered on the first day of an absolute neutrophil count above 0.5 × 109/L, platelet engraftment on the first of 5 subsequent days of platelet counts more than 50 × 109/L without platelet transfusions, as well as by full donor chimerism at day +30.
Graft failure included all cases of primary graft failure and graft rejection after demonstrated engraftment with the detection of more than 90% of the patient’s chimerism in peripheral blood or bone marrow.
GVHD and viral infections
Acute and chronic GVHD were diagnosed and graded using standard guidelines.12 Cases of acute GVHD grade 1 were treated with topical steroids/tacrolimus with no systemic steroid therapy.
For CMV and EBV detection in blood, intravitreal fluid, cerebrospinal fluid, and bronchoalveolar lavage quantitative real-time polymerase chain reaction analysis was used. CMV and EBV levels were routinely monitored weekly until CD4+ T-cell recovery was above 0.3 × 109/L. On detection of any grade of CMV viremia, patients received either ganciclovir or foscarnet (in patients with cytopenia), continued for at least 2 weeks after the last positive viremia test. CMV disease was identified with the detection of CMV in affected tissue. Routine ophthalmoscopic examination was performed in all patients with CMV viremia once every 2 weeks. CMV retinitis was diagnosed if a typical ophthalmoscopic picture was present, and in all patients, it was confirmed by polymerase chain reaction detection of CMV in the vitreal fluid. All but 1 patient received intraocular injection of antiviral drugs (ganciclovir or foscarnet) weekly before complete virus elimination from the vitreal fluid. CMV pneumonia and encephalitis were proven by the detection of CMV in the bronchoalveolar lavage or cerebrospinal fluid.
As combined PIDs are known to be predisposed to opportunistic infections, including CMV, we compared these patients vs patients with PID with a lower predisposition to viral infections before HSCT: defects of phagocytosis and immune dysregulation.1
Statistical analysis
Statistical analysis was performed in May 2018. Detailed information on statistical tests is represented in supplemental Data 2.
An undetected date of platelet engraftment in patients with thrombocytopenia, but full donor chimerism at day +30, was an exclusion criterion for platelet engraftment analysis. Four patients were excluded from the platelet engraftment analysis in the MUD group: 1 with primary graft failure, 1 who died of sepsis before platelet engraftment, and 2 with secondary thrombocytopenia with donor chimerism on day +30 after HSCT. Two patients were excluded from the MMRD group: 1 with primary graft failure and 1 with secondary thrombocytopenia and donor chimerism on day +30.
Four patients who received different doses of cyclophosphamide were excluded from the analysis of conditioning regimens influence on graft failure incidence. For the analysis of graft cells counts influence on graft failure incidence, all patients were divided into 2 groups: those who receive above and below the median level (nucleated cells [NC], 7.3 × 108/kg; CD34+, 10.9 × 106/kg; CD3+TCRαβ+, 16 × 103/kg).
The incidence of CMV reactivation was estimated in 94 patients. Four patients (3, WAS; 1, XLP type 2) were excluded because of active CMV disease at the time of HSCT.
Results
Engraftment and graft failure
Neutrophil engraftment occurred at a median of 14 days after HSCT in 74 patients in the MUD group (range, 9-28 days) and in 22 patients in the MMRD group (range, 9-20 days; P = .93). Platelet engraftment was achieved at a median 13 days after HSCT in 71 patients in the MUD group (range, 9-24 days) and in 21 patients after MMRD (range, 7-29 days; P = .43). One patient in each group (both with SCN) had primary graft failure. The cumulative incidence of graft failure was 0.16 (95% confidence interval [CI], 0.1-0.27) in the MUD group and 0.17 (95% CI, 0.07- 0.42) in the MMRD group (P = .85). Fourteen patients developed graft rejection with a median time of 2.8 months after HSCT (range, 1.0-6.9 months). The levels of NC, CD34+, and CD3+TCRαβ+ cells in graft did not significantly influence rates of graft failure (P = .12, .9, and .94, respectively). The only factor significantly influencing graft failure incidence was the conditioning type. In HSCT with 1 alkylating agent (n = 25), the graft failure incidence was 0.32 (95% CI, 0.18-0.57), which was significantly higher than that in HSCT with 2 alkylators (n = 52): 0.12 (95% CI, 0.06-0.25; P = .03; supplemental Figure 1A). None of the 17 patients with WAS who additionally received G-CSF with plerixafor during conditioning rejected the graft.
Acute and chronic GVHD
The cumulative incidence of acute GVHD grade II to IV was 0.17 (95% CI, 0.1-0.28) and 0.22 (95% CI, 0.1-0.47; P = .7) in the MUD and MMRD groups, respectively (supplemental Figure 2A). All but 2 patients had grade II GVHD with quick resolution of symptoms after receiving first-line therapy with systemic steroids. In both cases of grade III GVHD, it was associated with severe viral infections: adenovirus in blood and gut and human herpes virus 6 (HHV6) in blood and skin. It resolved with a combination of immunosuppressive and antiviral therapy. None of 2 patients with no serotherapy in preparative regimens developed GVHD. The cumulative incidence of chronic GVHD was 0.09 (95% CI, 0.04-0.19) in the MUD group and 0.13 (95% CI, 0.05-0.37) in the MMRD group (P = .96; supplemental Figure 2B). All 9 cases of chronic GVHD were limited and completely resolved with immunosuppressive therapy.
Viral reactivations
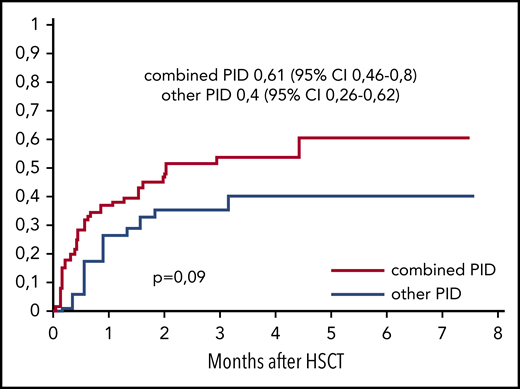
The cumulative incidence of CMV viremia was 0.5 (95% CI, 0.38-0.65) in the MUD group (n = 74) and 0.6 (95% CI, 0.42-0.86) in the MMRD group (n = 20; P = .35). The median time to CMV reactivation was 2.4 weeks after HSCT (range, 0.3-18.8 weeks). Posttransplant CMV disease occurred in 16 (17%) of 94 patients, and in 13 of 16 patients it manifested with retinitis. Three patients had pneumonia, and 1 had a combination of retinitis, pneumonia, and encephalitis. Importantly, of the patients who developed CMV disease, 14 had combined PID. A trend of higher incidence of CMV reactivation was observed in patients with combined PID (0.61; 95% CI, 0.46-0.8; n = 61) in comparison with patients with PID with defects of phagocytosis and immune dysregulation (0.4; 95% CI, 0.26-0.62; n = 34; P = .09; Figure 1).
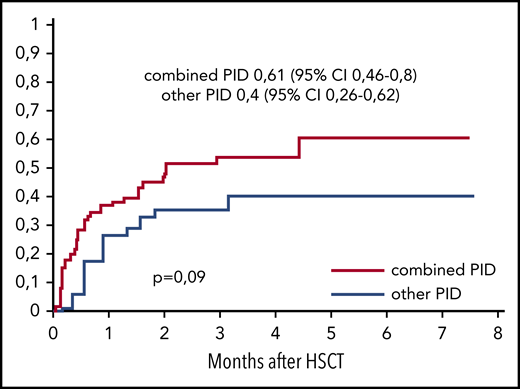
The cumulative incidence of CMV reactivation after HSCT from MUDs and MMRDs in patients with combined PIDs (n = 60) and other PIDs (n = 34).
The cumulative incidence of CMV reactivation after HSCT from MUDs and MMRDs in patients with combined PIDs (n = 60) and other PIDs (n = 34).
The probability of EBV viremia was 0.35 (95% CI, 0.23-0.52) in the MUD group and 0.09 (95% CI, 0.02-0.33) in the MMRD group (P = .09). We looked at the factors that could possibly lead to this difference. The first was preventive rituximab administration. Most patients (20 of 23) in the MMRD group received rituximab in the conditioning regimen, but the addition of rituximab in the MUD group did not significantly influence the incidence of EBV reactivation (P = .91). Second, we examined donor EBV serological statuses. Only 1 MMRD was EBV negative. Although 13 of 75 MUDs were EBV negative, EBV viremia occurred in only 2 patients after HSCT from EBV-negative MUDs. Thus, donor EBV status seemed to have no effect on the EBV reactivation rate.
There were no cases of posttransplant lymphoproliferative disease, and only 1 patient received preemptive therapy with rituximab.
Immune recovery
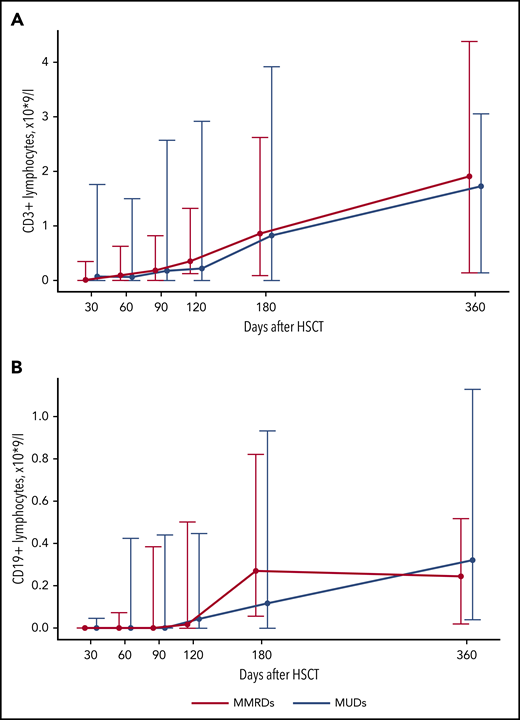
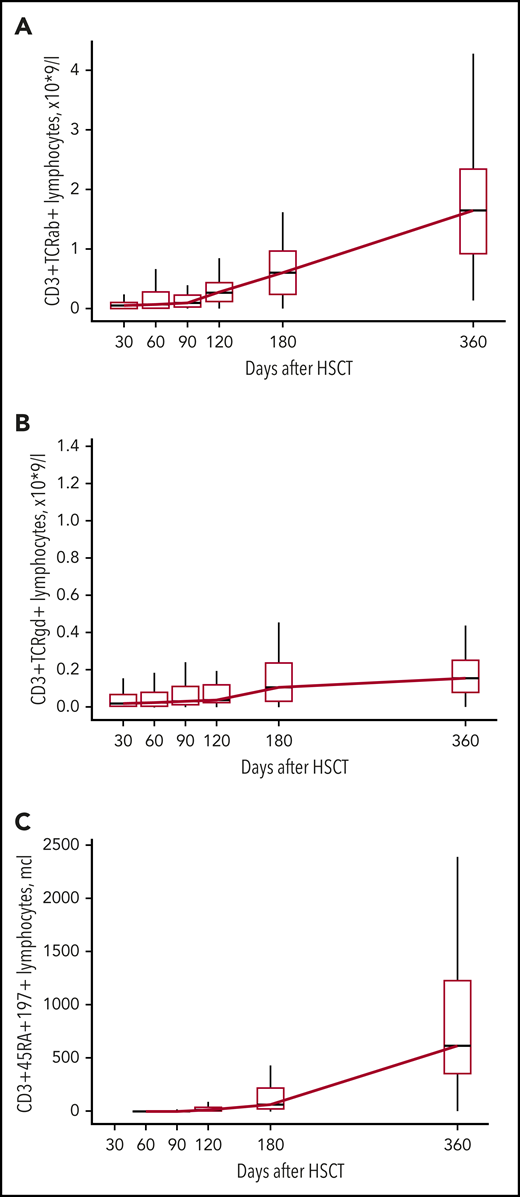
The increase in CD3+ cells in most patients was observed after day +120 and was mostly a result of the increase of CD3+TCRαβ+ cells (Figures 2A and 3A; supplemental Table 2). An increase in naive T cells (CD3+CD45RA+CD197+) was also observed at day +120 (Figure 3C; supplemental Table 2). CD3+TCRγδ+ cells were detected throughout the posttransplant period (Figure 3B; supplemental Table 2). The appearance of B cells was also observed after days +90 to +120 (Figure 2B; supplemental Table 2). There was no significant difference in the kinetics of CD3+ and CD19+ immune recovery between the MUD and MMRD groups (Figure 2A-B; supplemental Table 2), with a P value in the range 0.08 to 0.99 for CD3+ cells and 0.13 to 0.66 for CD19+ cells at the time after HSCT.
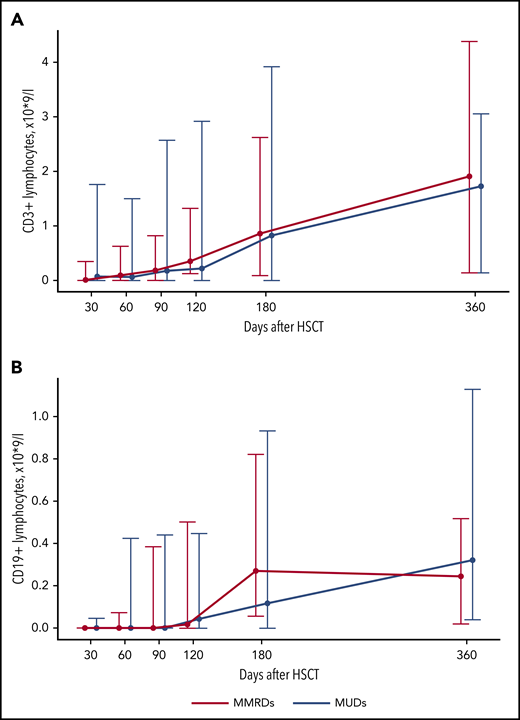
Kinetics of CD3+ and CD19+ lymphocyte recovery after HSCT from MUDs and MMRDs. Median-range graphs with medians, minimum and maximum of CD3+ (A) and CD19+ (B) lymphocyte counts (×109/L) at different time points after HSCT. P values for comparisons between MUD and MMRD groups at each time point are represented in supplemental Table 2.
Kinetics of CD3+ and CD19+ lymphocyte recovery after HSCT from MUDs and MMRDs. Median-range graphs with medians, minimum and maximum of CD3+ (A) and CD19+ (B) lymphocyte counts (×109/L) at different time points after HSCT. P values for comparisons between MUD and MMRD groups at each time point are represented in supplemental Table 2.
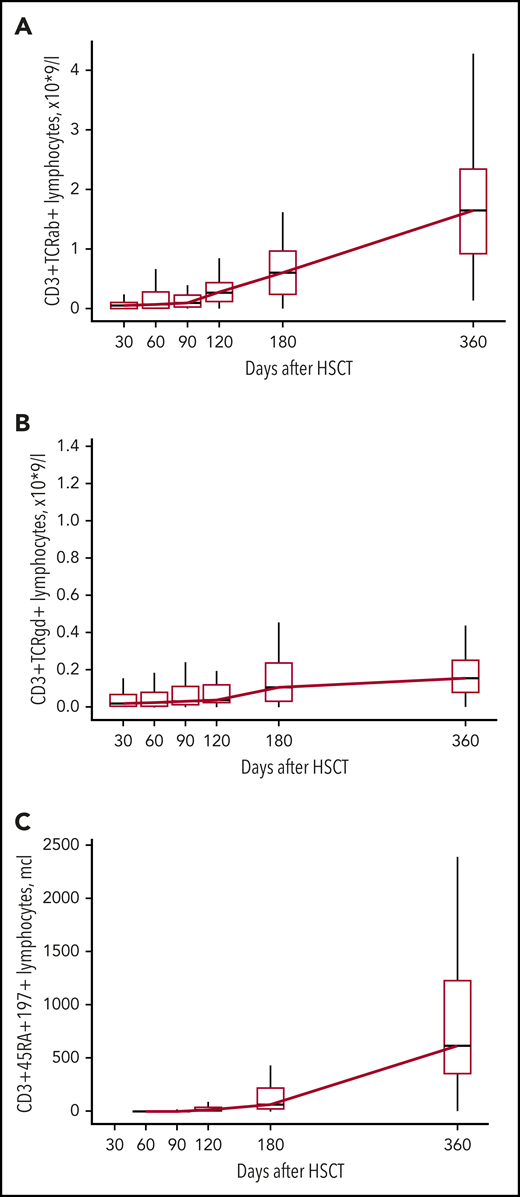
Kinetics of CD3+TCRαβ+, CD3+TCRγδ+, and CD3+CD45RA+CD197+ lymphocyte recovery. Boxplot graphs with medians, first and third quartiles of CD3+TCRαβ+ (A), CD3+TCRγδ+ (B), and CD3+CD45RA+CD197+ (C) lymphocyte counts (×109/L) at different time points after HSCT.
Kinetics of CD3+TCRαβ+, CD3+TCRγδ+, and CD3+CD45RA+CD197+ lymphocyte recovery. Boxplot graphs with medians, first and third quartiles of CD3+TCRαβ+ (A), CD3+TCRγδ+ (B), and CD3+CD45RA+CD197+ (C) lymphocyte counts (×109/L) at different time points after HSCT.
At a minimal 1-year FU after HSCT, 48 of 66 alive patients with functioning grafts were IV immunoglobulin (IVIG) independent. Eighteen patients required IVIG transfusions: 8 because of chronic GVHD, 2 patients with WAS because of predominantly recipient B-cell chimerism, 3 because of autoimmune phenomena, and 5 because of hypogammaglobulinemia. Only 2 patients with WAS with highly predominant recipient B-cell chimerism required IVIG transfusions 2 years after HSCT.
Survival and mortality rates
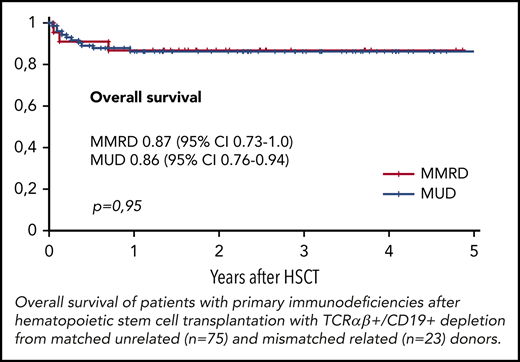
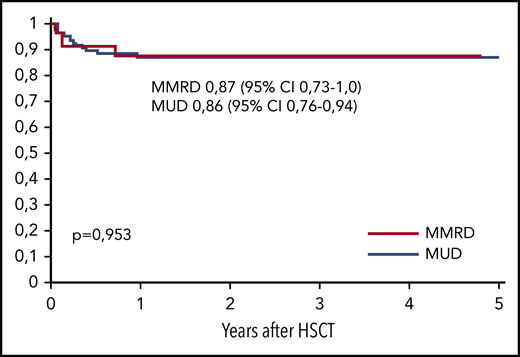
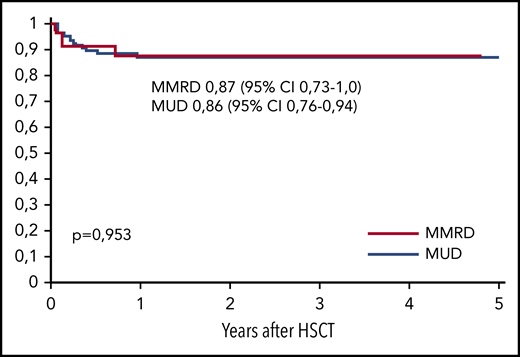
The overall survival (OS) was similar in both groups, at 0.86 (95% CI, 0.79-0.94) in the MUD group and 0.87 (95% CI, 0.73-1.0) in the MMRD group (P = .95; Figure 4). Thirteen of 98 patients died, 10 of whom died after the first HSCT and 3 of whom died after the second transplantation. The cause of death in all these patients was infection: 7 with Gram-negative sepsis and 6 with viral infection (adenoviral hepatitis, 2; CMV pneumonia, 4). One of 4 patients (with XLP type 2) developed CMV pneumonia before HSCT. Transplant-related mortality after first HSCT was 0.11 (95% CI, 0.06-0.21) in the MUD group and 0.09 (95% CI, 0.02-0.33) in the MMRD group (P = .81). The probability of OS was significantly lower in patients without therapeutic control of severe infectious and autoimmune complications at the moment of HSCT (0.52; 95% CI, 0.28-0.79) than in those who had no complications (0.93; 95% CI, 0.87-0.98; P < .0001; supplemental Figure 3).
At last FU time, all but 3 (2 WAS with predominately donor whole blood chimerism and no immunoglobulin production and 1 WAS with vision lost as a result of CMV retinitis) patients were free of prior disease symptoms and transplant-related end organ damage. Event-free survival was similar in MMRD (0.7; 95% CI, 0.51-0.88) and MUD (0.71; 95% CI, 0.6-0.81) groups (P = .86). In patients with conditioning regimens containing 2 alkylators and combination of G-CSF with plerixafor in WAS (n = 62), event-free survival was 0.84 (0.75-0.93).
Discussion
According to the European Society for Blood and Marrow Transplantation, the use of alternative donors for HSCT has increased dramatically within the last several years.13 The broad implementation of different T-cell depletion technologies has led to better outcomes of HSCT procedures from alternative donors. A variety of methods was used to remove alloreactive T lymphocytes from grafts. Along with immunomagnetic ex vivo cell selection that has been proven effective in patients with PID,4 the technology of in vivo T-cell depletion with posttransplant cyclophosphamide administration looks promising, although sufficient data in patients with PID is still lacking.14,15 TCRαβ+/CD19+ depletion is a method of graft engineering resulting in the removal of TCRαβ+ lymphocytes, which contain potentially recipient alloreactive T cells. Large amounts of TCRγδ+ and NK cells remaining in the graft are supposed to improve immune recovery and control infection.16 The current study presents the first comparative description of the results of HSCT with TCRαβ+/CD19+ graft depletion in a cohort of patients with various PIDs who underwent transplantation from MUDs or MMRDs.
For many years, HSCT in patients with PID from MMRDs has been associated with significantly lower OS that from MUDs and matched siblings.2 The current study demonstrates that the introduction of TCRαβ+/CD19+ graft depletion can potentially shift this tendency toward more favorable outcomes. In our experience, we observed a high survival rate (up to 0.87) after both MUD and MMRD transplantations. The factor that significantly and negatively influenced survival rate for our patients with PID was the lack of control of infectious and autoimmune complications at the time of transplantation. The OS in this group was only 0.52 compared with 0.93 in patients without severe complications at the time of HSCT.
Although graft T-cell elimination efficiently prevents GVHD after MMRD HSCT, the risks for life-threating GVHD after usage of unmanipulated grafts from MUDs in PIDs remain a serious issue.17 We demonstrated earlier favorable effects of TCRαβ+/CD19+ on GVHD prophylaxis in HSCT from both MMRDs and MUDs.6,8 In the current study, the rate of acute GVHD was low: only 0.17 in MUD and 0.22 in MMRD groups. In most cases, acute GVHD was limited to grade 2 and responded well to the first-line therapy. Only 2 patients had grade 3 acute GVHD, but in both cases, it was associated with significant viral infections. We also observed only limited forms of chronic GVHD with complete resolution after immunosuppressive therapy. None of the patients with GVHD experienced severe morbidity or died. The low incidence оf GVHD and absence of severe GVHD, in conjunction with the high incidence of viral infections after HSCT, makes abandoning posttransplant immunosuppressive therapy an attractive option. This approach was previously suggested by Bertaina et al,5 although formal proof of such an approach in larger cohorts of patients is needed.
T-cell elimination from grafts typically leads to increased risks for graft failure.4 Although the rate of primary graft failure in our study was relatively low, we observed a high incidence of secondary graft rejections. Interestingly, all cases of graft rejection occurred relatively early after HSCT, mostly within the first 6 months. Importantly, donor type, the levels of graft NC, CD34+ and CD3+TCRαβ+ cells did not affect the incidence of graft rejection. In fact, the only factor significantly influencing the incidence of graft failure was the intensity of the preparative regimen. In this study, we confirmed our previous results demonstrating an increased rate of graft rejection after a conditioning regimen with treosulfan as the single alkylating agent.6 The addition of a second alkylator significantly improved the results of engraftment, although late graft dysfunction remains an issue in some group of patients, such as those with WAS. To circumvent this problem, we used the combination of plerixafor and G-CSF, which appears to be a safe and effective approach.8 However, we evaluated the efficacy of conditioning regimens with a combination of plerixafor and G-CSF only in patients with WAS; hence, the advantages of mobilization need further confirmation in other types of PID. Importantly, changing the strategy of conditioning regimens with the addition of a second alkylating agent and a combination of G-CSF with plerixafor in WAS raised event-free survival in our group of PID after HSCT with TCRαβ+/CD19+ graft depletion from 0.7 to 0.86.
Swift immune recovery after HSCT is critically important for patients with PID because transplantation in these patients is usually aimed at fully controlling infections and autoimmunity. TCRαβ+/CD19+-depleted grafts contain immune competent cells, mostly natural killer and CD3+TCRγδ+ cells.18 In our study, as well as in the previous study by Airoldi et al.,16 persistent low numbers of CD3+TCRγδ+ early after HSCT were observed. A significant increase in the T-cell levels was observed after day +120. At this time, there is an elevation of CD3+TCRαβ+ and naïve (CD3+CD45RA+CD197+) T cells, which marks the appearance of thymus-derived cells, as confirmed by diversification of the TCR repertoire after day +120.19 There was no significant difference between the kinetics of immune reconstitution after MUD and MMRD HSCT.
Viral reactivation in patients with PID before HSCT is an essential issue unfavorably influencing the outcomes of transplantation.7 The incidence of CMV reactivation in our patient groups was comparable to the results of our previous study in TCRαβ+/CD19+-depleted HSCT for pediatric patients with various disorders without any difference between the MUD and MMRD groups.20 Interestingly, the median time of CMV reactivation in this study was 2 weeks after HSCT, whereas in patients with other diagnoses, it was 5 weeks,20 suggesting a presumably higher rate of CMV infection in patients with PID before HSCT. We also observed a trend toward a higher rate of CMV reactivation in patients with combined immune defects in comparison with other PIDs. The rate of CMV disease in our study was high (17%), but the predominant manifestation was CMV retinitis (14 of 16 cases). Posttransplant CMV retinitis seems to be common in patients with PID in comparison with patients with other disorders.21 Similar observations regarding the prevalence of retinitis as the most common manifestation of CMV disease were reported in a large cohort of individuals with HIV-driven immune deficiencies.22 Nevertheless, there are no protocols for CMV retinitis monitoring and management in HSCT recipients. In 1 of the first patients in our study (with WAS), CMV retinitis resulted in vision loss. Hence, we opted for regular ophthalmoscopic monitoring of patients with PID with posttransplant CMV viremia. Such an approach might have led to an overdiagnosis of CMV retinitis in our patients; therefore, further studies to design the correct strategy for CMV retinitis management in HSCT recipients are needed.
Despite the high expectations of the possible antiviral effects of TCRγδ+ cells infused with a graft, CMV reactivation remains a significant issue leading to morbidity and mortality in patients with PID after TCRαβ+/CD19+-depleted transplantations. The use of CMV-seronegative donors typically leads to increased risks for posttransplant CMV reactivation.23 For most patients with combined PID who are more likely to acquire CMV because of immune system defects, the CMV status before HSCT remains unknown because of the ambiguous interpretation of serology testing. The adoptive transfer of donor memory T cells (CD45RA-depleted) is a promising method for the prophylaxis and treatment of viral infections.24 Considering this recently, the strategy of donor searches for patients with combined PID in our center has been changed, with a preference of CMV-positive donors. The advantages of this technology are the multispecific effect of memory T cells against various types of infectious agents and the possibility of the adoption of immunological memory of parents (who are often exposed with common pathogens with their children and have more appropriate memory T lymphocyte repertoire) in MMRD transplantations. However, more detailed randomized studies of this approach are needed.
Our data confirm previous reports of adequate control of posttransplant EBV infections after B-lymphocyte elimination from grafts with CD19+ depletion and pretransplant rituximab administration.20
The most important conclusion from our current study is the absence of any significant differences in HSCT outcomes using TCRαβ+/CD19+-depleted grafts from MUDs and MMRDs. This suggests that TCRαβ+/CD19+ graft depletion is an effective and safe technology of HSCT in patients with PID receiving transplants from either MUDs or MMRDs. One can argue that TCRαβ+/CD19+ depletion is expensive. However, the cost of the depletion procedure is comparable to or less than the combined cost of a MUD search and procurement, along with potential treatment costs of severe refractory GVHD after unmanipulated MUD HSCT, which have been evaluated by pharmacoeconomic analysis.25
We conclude that in the absence of HLA-identical siblings, HSCT from MMRDs with TCRαβ+/CD19+ graft depletion is a safe and effective alternative for MUD HSCT in patients with PID. The availability of MMRD for most pediatric patients and the opportunity to easily perform a posttransplant donor to patient adoptive cell therapy are the advantages of MMRD use. Regarding the results of our study for HSCT with TCRαβ+/CD19+ graft depletion, donor HLA compatibility should not be considered an important factor influencing HSCT results for PID. The abovementioned benefits of MMRD should be considered as additional factors for donor choice.
For original data, please contact alexandra.laberko@gmail.com. Anonymous individual participant data that underlie the reported results will be made available 6 months after publication date at www.ClinicalTrials.gov. Proposals for access should be sent to bala8@yandex.ru.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the HSCT and Immunology Units and other hospital division staff, “Podari Zhizn” charitable fund for continued support of the transplantation program, Marina Persiantseva and Stefan-Morsch-Stiftung for years of collaboration in unrelated donors transplantation, and Rimma Khismatullina for the coordination of related donor transplantations.
Authorship
Contribution: A.L. collected, analyzed, and interpreted the data and prepared the manuscript; E.S. assisted with data collection; E.G., I.S., and L.S. contributed to HSCT therapy; E.K. performed related donor apheresis; Y.M. and A.K. performed graft processing; D.P. coordinated and performed flow cytometry for immune recovery analysis; K.V. contributed to statistical analysis; A.S. led pretransplant PID patient care; M.M., A.M., and D.B. led the HSCT program and edited the manuscript; and all authors read and approved the final manuscript.
Conflict-of-interest disclosure: M.M. received lecturer’s fees from Miltenyi Biotec. The remaining authors declare no competing financial interests.
Correspondence: Alexandra Laberko, Department of Immunology, Dmitry Rogachev National Medical Center of Pediatric Hematology, Oncology and Immunology, Samory Mashela Str, 1, 117997, Moscow, Russia; e-mail: alexandra.laberko@gmail.com.