

Key Points
Concizumab was safe and well tolerated in HA and HAwI/HBwI, including with breakthrough bleed treatment.
Clinical proof of concept in terms of preventing bleeds was demonstrated in 2 concizumab phase 2 trials across hemophilia subtypes.

Abstract
Results from the main parts (24 weeks) of 2 concizumab phase 2 trials are presented: explorer4 in hemophilia A (HA) or B (HB) with inhibitors (HAwI/HBwI) and explorer5 in HA. The trials aimed to evaluate the efficacy of daily subcutaneous concizumab prophylaxis (evaluated as annualized bleeding rate [ABR] at last dose level), with secondary objectives being safety and immunogenicity (assessed as number of adverse events [AEs] and antidrug antibodies [ADAs]). Patients received 0.15 mg/kg concizumab, with potential dose escalation to 0.20 and 0.25 mg/kg (if ≥3 spontaneous bleeding episodes within 12 weeks of concizumab treatment). Relevant pharmacokinetic/pharmacodynamic (PK/PD) parameters were assessed. Thirty-six HA, 9 HAwI, and 8 HBwI patients were exposed to concizumab. Most inhibitor patients (15 of 17; 88.2%) did not escalate the dose; all patients chose to continue to the extension phase of the trials. Clinical proof of concept for prevention of bleeding episodes was demonstrated in both trials. Estimated ABRs in HAwI and HBwI were lower vs HA: 3.0 (95% confidence interval [CI], 1.7; 5.3) and 5.9 (95% CI, 4.2; 8.5) vs 7.0 (95% CI, 4.6; 10.7), respectively. PK/PD results were as expected, with no difference between hemophilia subtypes for concizumab exposure, free tissue factor pathway inhibitor, thrombin generation, prothrombin fragment 1+2, and d-dimers. Concizumab was safe and well tolerated (no severe AEs, AE-related withdrawals, or thromboembolic events). Three patients had (very low to medium titer) ADA+ tests in each trial, with no observed clinical effect. These results support further development of concizumab as a daily prophylactic treatment in all hemophilia patients. These trials were registered at www.clinicaltrials.gov as #NCT03196284 and #NCT03196297.
Introduction
Patients with congenital hemophilia exhibit an increased bleeding tendency as a result of deficiency/absence of coagulation factor VIII (FVIII; hemophilia A [HA]) or factor IX (FIX; hemophilia B [HB]) activity.1 Prophylactic treatment with the missing coagulation factor is the current recommended standard of care in hemophilia to prevent fatal bleeding and bleeding-related complications.2 Current prophylactic regimens are associated with a significant burden of care due to the need for frequent IV infusions.3-6 Even with the advent of new, extended half-life FVIII and FIX products allowing for less frequent infusions, reconstitution and IV administration are still required.7 Moreover, recent real-world data have highlighted that a significant proportion of patients do not achieve the desired outcomes with current therapies.8 Novel subcutaneous nonreplacement treatments are in clinical development, with 1 (emicizumab [Hemlibra]; F. Hoffmann-La Roche AG, Basel, Switzerland) recently approved for patients with HA with inhibitors (HAwI) or without inhibitors.
The development of antibodies (inhibitors) against exogenous FVIII or FIX that render factor replacement therapy ineffective represents the most common and challenging treatment-associated complication in people with hemophilia. Immune tolerance induction (ITI) is the standard treatment for the management and eradication of such inhibitors, but this procedure fails to achieve tolerance in a substantial number of patients.9 Patients with inhibitors can be treated with bypassing agents (activated recombinant factor VII [rFVIIa]; activated plasma-derived prothrombin complex concentrate [pd-aPCC]); however, administration may be burdensome, response to treatment variable, and the efficacy profile of these agents is generally inferior to replacement therapy.9,10 Patients with HAwI can also use emicizumab, although there may be some thrombotic risk associated with pd-aPCC use to treat breakthrough bleeding episodes.11 Historically, fewer HB with inhibitor (HBwI) patients have responded to ITI than HAwI patients and may develop unusual events related to FIX exposure, including anaphylactoid reactions or nephrosis; their treatment options are generally limited to on-demand rFVIIa/pd-aPCC treatment. In addition, a significant proportion of patients with inhibitors receive delayed or suboptimal treatment and their management remains a major challenge.12 Overall, unmet needs remain in hemophilia patients for safe and effective therapeutic agents that can be administered subcutaneously in the prophylactic setting.13,14
Concizumab, a high-affinity, humanized, anti–tissue factor (TF) pathway inhibitor (TFPI) monoclonal antibody, is in clinical development for the subcutaneous treatment of patients with HA, HB, HAwI, and HBwI. TFPI is a potent inhibitor of the coagulation initiation phase, more specifically the activation of factor X (FX) to FXa by the TF/FVIIa complex. TFPI first binds to and inhibits FXa and subsequently binds to and inhibits TF/FVIIa, forming a TF/FVIIa/FXa/TFPI complex.15,16 Therefore, concizumab prevents both inhibition of FXa and inhibition of FVIIa/TF by TFPI, thereby allowing generation of sufficient amounts of FXa to ensure effective hemostasis in the absence of a functional activated FIX/activated FVIII (FIXa/FVIIIa) complex in patients with hemophilia.16-19 In a phase 1b trial, the pharmacodynamic (PD) relationship between concizumab exposure, free TFPI, and thrombin generation (TG) was confirmed.20
In this manuscript, we present results from the main parts (24 weeks) of 2 phase 2 trials that assessed the efficacy and safety of subcutaneous concizumab administration in patients with HAwI/HBwI (explorer4) and in patients with severe HA without inhibitors (explorer5). The studies also aimed to establish an optimal dose for the phase 3 concizumab trials. The designs of the 2 trials were sufficiently similar to allow evaluation of data across the 2 studies, specifically in terms of comparison by hemophilia subtype, and results are therefore presented together.
Materials and methods
Trial design
explorer4 (inhibitor trial) was a multicenter, open-label, randomized controlled trial, conducted at 17 sites in 12 countries. explorer5 (noninhibitor trial) was a multicenter, single-arm, open-label phase 2 trial, conducted at 26 sites in 11 countries. Both trials were approved according to local regulations by the appropriate health authorities and by an independent ethics committee or institutional review board, as required, and conducted in accordance with the Declaration of Helsinki and International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Good Clinical Practice. Written informed consent was obtained from all participants prior to any trial-related activities.
The dosing regimen was selected based on phase 1 pharmacokinetic (PK)/PD results, with dosing commencing at the lowest possible dose to ensure that two-thirds of patients would achieve concizumab steady-state concentrations >100 ng/mL, as it had been shown that the TG potential was reestablished to within the normal range and there was a decrease in reported bleeding episodes at those concentrations.21 Patients in both trials were treated with daily subcutaneous injections of 0.15 mg/kg concizumab, with potential dose escalation to 0.20 and 0.25 mg/kg if they experienced ≥3 spontaneous bleeding episodes within the preceding 12 weeks of concizumab treatment (Figure 1). In the inhibitor trial, a loading dose of 0.5 mg/kg concizumab was administered as the first dose. A loading dose was not given in the noninhibitor trial. Cautious dose escalation was implemented to allow for selection of a dose that was efficacious and safe for each patient. In the inhibitor trial, patients were randomized 2:1 to prophylaxis with concizumab or on-demand treatment with eptacog alfa activated (rFVIIa; NovoSeven; dosed at the investigators’ discretion). The loading dose, followed by 1 week of concizumab dosing (0.15 mg/kg), was given to ensure steady-state levels at the time of the first rFVIIa administration and to assess potential safety issues. Randomization was performed using an interactive web-response system. Breakthrough bleeding episodes occurring between the first administration of concizumab and the end-of-trial visit were treated at home. Noninhibitor patients treated bleeding episodes with nonmodified (standard half-life) FVIII (either NovoEight, provided by Novo Nordisk, or another nonmodified FVIII product that was not provided by Novo Nordisk) at the discretion of the investigator, as long as any given single dose did not exceed 50 IU/kg and inhibitor patients treated breakthrough bleeding episodes with rFVIIa (≤90 μg/kg at the investigators’ discretion; provided by Novo Nordisk). In retrospect, the omission of a loading dose in the noninhibitor trial was recognized as a weakness in the trial design, which will be addressed in the upcoming phase 3 trials (in which a loading dose will be administered to all patients; see also “Discussion”).

Study design for the phase 2 concizumab trials. (A) explorer5 (HA without inhibitors) and (B) explorer4 (HAwI/HBwI). Dose-escalation criteria: if a patient experienced ≥3 spontaneous bleeding episodes within the preceding 12 weeks of treatment with concizumab, the patient could be escalated to the next dose level. In explorer4, after completion of the main part, patients in the rFVIIa arm continuing on to the extension part of the trial were switched to daily prophylactic subcutaneous treatment with concizumab 0.15 mg/kg, with potential dose escalation as appropriate. ↑, Dose escalate to the next dose level, based on the dose-escalation criteria. R, randomization.
Study design for the phase 2 concizumab trials. (A) explorer5 (HA without inhibitors) and (B) explorer4 (HAwI/HBwI). Dose-escalation criteria: if a patient experienced ≥3 spontaneous bleeding episodes within the preceding 12 weeks of treatment with concizumab, the patient could be escalated to the next dose level. In explorer4, after completion of the main part, patients in the rFVIIa arm continuing on to the extension part of the trial were switched to daily prophylactic subcutaneous treatment with concizumab 0.15 mg/kg, with potential dose escalation as appropriate. ↑, Dose escalate to the next dose level, based on the dose-escalation criteria. R, randomization.
Results from the main portions of both trials are presented here. For the inhibitor trial, the results presented reflect data up to the last scheduled visit prior to and including 19 September 2018.
For the noninhibitor trial, the results presented reflect the data up to the last scheduled visit prior to and including 22 June 2018. The duration of treatment of the main portion was at least 24 weeks for those receiving concizumab (ie, the cutoff date occurred when the last patient had completed 24 weeks of treatment). For inhibitor patients randomized to on-demand treatment with rFVIIa, only data on the 24 weeks of on-demand treatment were included in the main portion analysis. For both trials, after 24 weeks, patients continued into the extension portion.
Trial population
Male patients, aged ≥18 years old, with HAwI/HBwI and documented history of high-titer (≥5 Bethesda units) inhibitors (explorer4) or with severe HA (FVIII activity <1%) without inhibitors (explorer5) were eligible to participate. HB patients without inhibitors did not participate in the noninhibitor trial as the effect of concizumab in this population is predicted to be similar to that in HA patients without inhibitors, and in order to avoid delays in recruitment and thereby completion of the trial (due to the limited availability of these patients for participation in clinical trials). Inhibitor patients who had been treated on demand with a minimum of 6 bleeding episodes during the 24 weeks (or 12 bleeds during 52 weeks) prior to screening were eligible. Noninhibitor patients who had previously been on prophylaxis or on-demand treatment were eligible; on-demand patients were required to have had a minimum of 6 bleeding episodes during the 24 weeks (or 12 bleeds during 52 weeks) prior to screening. Exclusion criteria are provided in supplemental Methods (available on the Blood Web site).
Objectives, end points, and assessments
The primary objective (both trials) was to assess the efficacy of once-daily, subcutaneous concizumab administration in preventing bleeding episodes in patients with HAwI/HBwI (explorer4) and in patients with severe HA without inhibitors (explorer5), evaluated as the number of bleeding episodes during at least 24 weeks from treatment initiation. All bleeding episodes were captured by patients in an electronic diary or by investigators in an electronic case report form. Bleeding episodes were evaluated based on cause (spontaneous, traumatic), location, and severity according to World Federation of Hemophilia guidance.2
Secondary objectives (both trials) included the assessment of concizumab safety and immunogenicity, evaluated as the number of treatment-emergent adverse events (TEAEs) and the occurrence of concizumab antidrug antibodies (ADAs) during at least 24 weeks from treatment onset, respectively. In patients with inhibitors, the safety of administering rFVIIa when exposed to concizumab was also a secondary objective and was assessed as the number of TEAEs within 24 hours of rFVIIa administration. Dosing of rFVIIa in the first 4 inhibitor patients randomized to the concizumab arm was staggered until the fourth randomized patient had completed visit 3 (at which rFVIIa was administered in a controlled setting) without safety concerns.
Assessment of concizumab ADAs was performed by specialized laboratories with a bridging electrochemiluminescence assay (binding ADA assay), using labeled concizumab for antibody capture and detection. ADA+ samples were further characterized for neutralizing activity using a modified TFPI functionality chromogenic assay (see supplemental Methods for more details).
Safety assessments also comprised clinical laboratory tests, including measurement of coagulation-related parameters such as prothrombin fragment 1+2 (F1+2) and d-dimers,20 physical examination, and vital signs. With the exception of hematology and urinalysis samples, which were analyzed locally, all other study laboratory analyses were performed by a central laboratory. In addition, prothrombin time, activated partial thromboplastin time, and fibrinogen measurements were performed locally at visit 3 in inhibitor patients.
Statistical analysis
Data were analyzed using SAS version 9.4 software. The sample size determined aimed to achieve an acceptably narrow 95% confidence interval (CI) for the annualized bleeding rate (ABR) in the noninhibitor trial and for the difference (in terms of ABR) between treatment arms in the inhibitor trial.
The full analysis set and the safety analysis set consisted of all dosed patients. All patients were included in both analysis sets.
The primary efficacy end point (ABR) in the primary analysis was evaluated at the last dose level reached for a patient; hence, only observations from the period during which patients were on their last dose level at the time of analysis were included in the analysis. Comparisons were made using a negative binomial model with log of exposure time at last dose level as offset. The 2-week run-in period for patients on 0.15 mg/kg concizumab at the time of analysis was excluded. In the inhibitor trial, the regimen (concizumab vs on demand) was a factor in the model, and proof of concept was concluded if the 95% CI of the treatment ratio was <1. In the noninhibitor trial, proof of concept was concluded if the 95% CI of the estimated ABR was <12. The value of 12 bleeds per year conservatively reflects a 50% reduction from the bleeding rate during on-demand treatment.
PK/PD end points were evaluated on the last concizumab dose level for patients having 0.15 mg/kg as the last dose level and presented by hemophilia type. Coagulation-related laboratory parameters were evaluated on last dose level and presented by hemophilia type. Safety end points are reported according to the actual dose level the patients were administering when they experienced the adverse event (AE)/at time of measurement. All other data are presented using descriptive statistics.
Results
Trial population and baseline characteristics
A total of 36 noninhibitor patients were enrolled and 32 completed the main portion (2 patients withdrew their consent; 1 withdrew from treatment; 1 withdrew as he experienced a lack of efficacy [there was no evidence of ADAs up to the time of withdrawal in these patients; see also supplemental Figure 1]). A total of 26 inhibitor patients were randomized to either concizumab (n = 17) or rFVIIa (n = 9), and 25 completed the main portion of the trial (1 patient in the rFVIIa arm withdrew his consent) (supplemental Figure 1). Baseline characteristics for inhibitor patients by treatment arm are shown in supplemental Table 1. Baseline characteristics for the patients who received concizumab in both trials are shown in Table 1 by hemophilia type. HA, HAwI, and HBwI patients had comparable baseline characteristics.
In the 12 months prior to screening, the majority of HA patients reported being on prophylactic treatment and had a lower bleeding rate than the HA patients who were being treated on demand (Table 1). All patients with inhibitors who received concizumab were treated on demand during the 12 months prior to screening, with 1 HAwI and 1 HBwI patient also reporting receiving prophylactic treatment during a short portion of that period. The highest on-demand mean ABRs prior to study initiation were reported among HBwI patients.
Efficacy in the explorer4 (inhibitor) and explorer5 (noninhibitor) trials
Concizumab dose escalation
Among HA patients, 21 (58.3%) remained on the initial concizumab dose (0.15 mg/kg), whereas 7 (19.4%) escalated to the 0.20 mg/kg and 8 (22.2%) to the 0.25 mg/kg dose levels. Among inhibitor patients, most (15 of 17; 88.2%) of those randomized to concizumab maintained the 0.15 mg/kg dose, whereas 2 (2 of 17; 11.8%) escalated to 0.20 mg/kg (Table 2).
Bleeding episodes
In the inhibitor trial, when assessing each patient’s last dose level, 47 treated bleeding episodes in 15 patients (88%) were reported in the concizumab prophylaxis arm in the main part of the trial, with an estimated ABR of 4.5 (95% CI, 3.2; 6.4) vs 20.4 (95% CI, 14.4; 29.1) in the rFVIIa on-demand arm (median ABR, 4.5 and 19.7 for concizumab and rFVIIa, respectively) (Figure 2A). The ABR ratio between the 2 treatment arms was 0.22 (95% CI, 0.13; 0.36), and as it was <1, clinical proof of concept was demonstrated. The proportion of spontaneous and traumatic bleeds in the concizumab arm was similar (51% vs 49%), and the estimated spontaneous ABR was 2.3 (95% CI, 1.4; 3.6). Most bleeds (72%) occurred in joints, and the estimated joint ABR was 3.2 (2.2; 4.7). None of the bleeds were characterized as severe.

ABRs. ABRs (bleeding episodes on last dose level) during the main part in (A) explorer4 (HAwI/HBwI), (B) explorer5 (HA without inhibitors), and (C) by hemophilia type in explorer4 and explorer5 (HA, HAwI, HBwI). ABRs were calculated based on a negative binomial regression model with log of exposure time in the main part of the trials as offset and treatment arm as factor. (A) Bleed reduction for treatment with rFVIIa vs concizumab was 78%, 88%, and 79% for all treated bleeds and for spontaneous and joint bleeds, respectively; ***P < .001. (B) Observations from the 2-week run-in are not included. In explorer4, there was 1 medication error in which a patient unintentionally received 5 × 15 mg/kg concizumab.
ABRs. ABRs (bleeding episodes on last dose level) during the main part in (A) explorer4 (HAwI/HBwI), (B) explorer5 (HA without inhibitors), and (C) by hemophilia type in explorer4 and explorer5 (HA, HAwI, HBwI). ABRs were calculated based on a negative binomial regression model with log of exposure time in the main part of the trials as offset and treatment arm as factor. (A) Bleed reduction for treatment with rFVIIa vs concizumab was 78%, 88%, and 79% for all treated bleeds and for spontaneous and joint bleeds, respectively; ***P < .001. (B) Observations from the 2-week run-in are not included. In explorer4, there was 1 medication error in which a patient unintentionally received 5 × 15 mg/kg concizumab.
In the noninhibitor trial, when assessing each patient’s last dose level, a total of 70 treated bleeding episodes in 23 patients (63.9%) were reported in the main part of the trial, with an estimated ABR of 7.0 (95% CI, 4.6; 10.7) and a median ABR of 4.5 (Figure 2B). Clinical proof of concept was also demonstrated for this trial, as the 95% CI for the ABR was <12. There was a lower proportion of spontaneous vs traumatic bleeds (37% vs 61%), and the estimated spontaneous ABR was 2.5 (95% CI, 1.5; 4.3). Most bleeds (63%) occurred in joints, and the estimated joint ABR was 4.9 (95% CI, 2.8; 8.5). A total of 3 bleeding episodes were assessed as severe.
Across the 2 trials, HA patients had the highest ABR estimate, as well as the highest joint ABR estimate of the 3 subtypes, with overlapping CIs (Figure 2C). The spontaneous ABR estimate was similar across the 3 hemophilia subtypes (Figure 2C).
ABRs for inhibitor patients randomized to receive rFVIIa on demand were comparable to historical ABRs. In contrast, inhibitor patients randomized to concizumab prophylaxis experienced reduced ABRs compared with historical values. Similarly, noninhibitor patients who escalated to higher concizumab doses in general treated fewer bleeding episodes per time on each dose level (supplemental Figure 2). However, it is not possible to determine to which extent this observation was simply due to uneven distribution of bleeds over time or a result of increased concizumab exposure.
PK and PD
Concizumab exposure was measured as the concentration in plasma over time. Concizumab concentration varied considerably among patients at the same dose level in each trial, as measured at week 24 (Table 3). Similar levels of concizumab exposure were observed in all 3 patient populations (Figure 3A). Because only 2 inhibitor patients escalated to 0.20 mg/kg concizumab (and none to 0.25 mg/kg), comparisons across hemophilia subtypes are shown for the patients who remained on 0.15 mg/kg concizumab (Figure 3A-C).
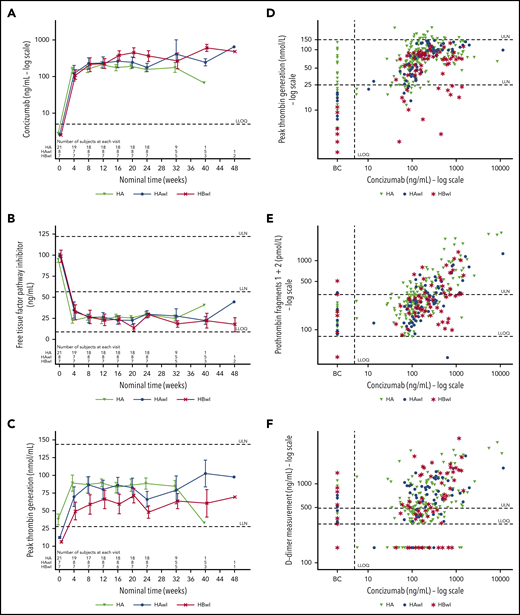
Mean plots. Mean plots of (A) concizumab plasma concentration vs time; (B) free TFPI vs time; (C) peak TG potential vs time; (D) peak TG potential vs concizumab plasma concentration; (E) PF1+2 vs concizumab plasma concentration; and (F) d-dimers vs concizumab plasma concentration by hemophilia type for patients with last dose level of concizumab 0.15 mg/kg (A-C) and for all dose levels (D-F) in the main parts of explorer4 (HAwI/HBwI) and explorer5 (HA without inhibitors). Baseline free TFPI, mean (standard deviation): 96.3 (11.1) ng/mL. Normal range of peakTG (lower; upper): 26; 147 nmol/L. BC, baseline values for concizumab arm; LLN, lower limit of normal; LLOQ, lower limit of quantification; ULN, upper limit of normal.
Mean plots. Mean plots of (A) concizumab plasma concentration vs time; (B) free TFPI vs time; (C) peak TG potential vs time; (D) peak TG potential vs concizumab plasma concentration; (E) PF1+2 vs concizumab plasma concentration; and (F) d-dimers vs concizumab plasma concentration by hemophilia type for patients with last dose level of concizumab 0.15 mg/kg (A-C) and for all dose levels (D-F) in the main parts of explorer4 (HAwI/HBwI) and explorer5 (HA without inhibitors). Baseline free TFPI, mean (standard deviation): 96.3 (11.1) ng/mL. Normal range of peakTG (lower; upper): 26; 147 nmol/L. BC, baseline values for concizumab arm; LLN, lower limit of normal; LLOQ, lower limit of quantification; ULN, upper limit of normal.
Overall, increasing concizumab exposure was associated with lower free TFPI and normalized TG potential. Corresponding to the increase in concizumab exposure, free TFPI was reduced to a similar extent across the different hemophilia subtypes when comparing patients exposed to 0.15 mg/kg concizumab (Figure 3B).
Peak TG potential at 24 weeks was within the normal reference range. Although peak TG potential was normalized across all 3 hemophilia subtypes, HBwI patients had lower levels of TG compared with HA and HAwI patients (Figure 3C), however, this is likely due to the effect of endogenous FIX in the TG assay that was used. Concizumab exposure was associated with increased TG potential (Figure 3D) and elevated F1+2 (Figure 3E) and d-dimers (Figure 3F), reflecting the hemostatic activity of TFPI inhibition. D-dimers seemed to increase with increasing concizumab concentration (baseline−week 24 mean change, 348.1 ng/mL [noninhibitor trial] and 265 ng/mL [inhibitor trial] in the concizumab arm); however, elevated baseline D-dimer levels were measured in both arms in the inhibitor trial. F1+2 was shown to increase with increasing concizumab plasma concentration (baseline−week 24 mean change, 271 pmol/L [noninhibitor trial] and 155 pmol/L [inhibitor trial] in the concizumab arm). F1+2 increases were, as expected, not observed with on-demand rFVIIa treatment. d-dimer and F1+2 levels fluctuated for the majority of patients during both trials. No significant changes in fibrinogen level or platelet counts were observed.
Safety
AEs
Overall, concizumab treatment was safe and well tolerated across all 3 dose levels in both trials. AE rates were low, with no severe AEs reported, no AE-related withdrawals, no thromboembolic events, and no deaths (Table 4). The majority of AEs in the concizumab arms were mild, unlikely related to concizumab and were resolved by the analysis cutoff date. There was 1 serious AE (SAE) (central venous catheter removal) reported in 1 inhibitor patient (6%) in the 0.15 mg/kg concizumab arm (Table 4), which was not assessed as possibly or probably related to concizumab. No AEs were reported within 24 hours following coadministration of concizumab and rFVIIa in inhibitor patients, and no safety issues were identified in association with the treatment of breakthrough bleeding episodes with rFVIIa. Similarly, there were no safety issues reported with the use of FVIII to treat breakthrough bleeding in noninhibitor patients.
Among the most frequently reported AEs in HA patients were nasopharyngitis (0.7 events per patient-years of exposure [PYE]); injection site bruising, hematoma, or hemorrhage (0.5, 0.3, or 0.4 events per PYE, respectively); and upper respiratory tract infection (0.1 events per PYE) (Table 4). There was 1 patient with injection site hematoma (0.2 events per PYE) and 1 with upper respiratory tract infection (0.3 events per PYE) among HAwI patients. Nasopharyngitis, injection site hematoma, and injection site hemorrhage (0.5, 0.9, and 0.9 events per PYE, respectively) were reported most frequently among HBwI patients (Table 4). All injection site reactions were mild, with the exception of 1 hemorrhage event and 1 event of ecchymosis, which were of moderate severity; both had resolved by the time of database lock.
Immunogenicity
Three patients in each of the 2 trials had positive ADA tests, with titers that were very low (1-16 in the noninhibitor trial) or medium (1-128 in the inhibitor trial), considering that the assay sensitivity is <1 ng/mL. One of the 3 noninhibitor patients and 2 of the 3 inhibitor patients had ADAs that were found to be neutralizing in vitro on 1 occasion, but not in any subsequent measurements. No apparent significant changes in bleeding pattern, PK/PD parameters, AEs, or laboratory coagulation-related parameters were observed in association with antibody development when compared with patients without ADAs.
Discussion
The phase 2 concizumab trials sought to assess the efficacy and safety of daily subcutaneous concizumab prophylaxis. A conservative concizumab dose escalation was chosen in the phase 2 trials due to a trend for a decrease in fibrinogen observed at the highest dose level in the phase 1b explorer3 trial.20 The studies included a heterogeneous group of hemophilia patients in terms of their treatment regimens and experiences prior to participating in the trials and results should be viewed in this context. Overall, concizumab was safe, and clinical proof of concept was demonstrated for both trials, indicating concizumab’s potential to exert prophylactic bleed protection across different hemophilia subtypes.
Mean ABRs were comparable to those observed during factor treatment in patients with and without inhibitors. Results from the inhibitor trial also confirmed a statistically significant (P < .001) and clinically relevant reduction in ABR compared with on-demand treatment (Figure 2A), and inhibitor patients randomized to concizumab prophylaxis showed a reduction in ABR compared with their historical ABRs (supplemental Figure 2A).
More HA patients escalated to the higher concizumab doses and their ABR estimate was higher compared with HAwI and HBwI patients, with overlapping CIs. A few HA patients in the noninhibitor trial had very high historical ABRs despite being on prophylaxis prior to study initiation (Table 1). It is conceivable that such patients may require concizumab levels higher than those to which they were exposed early in the trial, which did not include a concizumab loading dose (unlike the inhibitor trial) and did not allow a continuation of FVIII prophylaxis during the period prior to reaching concizumab levels predicted to be efficacious. In addition, because patients treated on demand in general bleed more frequently than those on prophylaxis, some of the patients in this group may have been able to better differentiate bleeding from pain or other symptoms associated with hemophilia than patients on prophylaxis. Consistent with this explanation, patients historically treated on demand in the noninhibitor trial exhibited a reduced bleeding rate. Importantly, all patients who completed the main parts of both trials chose to continue on to the extension, potentially reflecting a perceived prophylactic effect afforded by concizumab, as well as the reduced treatment burden associated with subcutaneous administration.22 This supports the assumption that avoiding the need for IV administration can potentially lead to better adherence, in turn allowing more patients to receive prophylactic treatment, and therefore contributing to improved outcomes.23
Treatment with concizumab in both phase 2 trials was associated with a favorable safety profile. There were no AE-related withdrawals, no deaths, and no thromboembolic events; most AEs were mild and unlikely related to concizumab. In addition, there were no safety issues related to concomitant use of rFVIIa or FVIII to treat breakthrough bleeds. With the exception of 2 moderate injection site reactions, all were mild and had recovered at database lock. Overall, no difference in AEs or laboratory safety markers was observed between HA, HAwI, and HBwI patients in the 2 trials.
Although not as crucial as the development of inhibitors with factor replacement therapy, the development of ADAs represents a significant complication for new therapies, as recently highlighted.24,25 In the concizumab phase 2 trials, a total of 6 patients had positive ADA tests, and 3 among them had antibodies that were neutralizing in vitro (at a single time point in each patient). Importantly, antibody development was not associated with a change in clinical status or observed concizumab efficacy in these patients.
The PK/PD results were as expected, consistent with phase 1 results20 and reflective of the hemostatic activity of TFPI inhibition, with no notable difference observed for concizumab exposure, free TFPI, TG, F1+2, and d-dimers between HA, HAwI, and HBwI patients, allowing extrapolation of these results to all patient subgroups. The lower TG potential observed in HBwI patients compared with HA and HAwI patients (Figure 3C-D) can be explained by the effect of endogenous FIX in the TG assay that was used (calibrated automated thrombogram; Thrombinoscope BV, Maastricht, the Netherlands). The influence of other factors on d-dimer levels (eg, infection or bleeding episodes) cannot be excluded, as this is a nonspecific thrombosis marker, and the elevated baseline d-dimer levels observed in the noninhibitor trial should be taken into consideration when assessing their increase following concizumab exposure. As previously noted, there were no thromboembolic events.
The concizumab phase 2 trial results have contributed important information, which will be applied to the design of 2 confirmatory phase 3 trials to determine optimal prophylactic efficacy without compromising safety. The proposed dosing regimen for the phase 3 trials will include a loading dose on the first day, which will serve to increase exposure during the initial trial period and reach steady-state levels faster, followed by a maintenance dose with the highest dose used in the phase 2 trials (0.25 mg/kg), administered as daily, subcutaneous injections from the second day onward. Results from both phase 2 trials presented here show that a maintenance dose of 0.15 mg/kg is suboptimal for some patients, as few (13%) experienced zero bleeds. When the ABR for each patient in the phase 2 trials was calculated based on their final dose levels, the 0.25 mg/kg dose appeared to provide the most optimal efficacy while maintaining safety (supplemental Figure 2), therefore, this will be the dose used in the planned phase 3 trials.
In conclusion, phase 2 trial results support the use of concizumab as a safe and well-tolerated subcutaneously administered prophylactic therapy in all patients with hemophilia, regardless of inhibitor status, and including patients with HBwI who represent a vulnerable and particularly rare patient population, and for whom the use of a novel agent could significantly improve care.
The subject-level analysis data sets for the research presented in the publication are available from the corresponding author on reasonable request.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients who participated in the trial and their families. The authors acknowledge the contribution of all investigators, coinvestigators, and study nurses who were involved in the explorer4 and explorer5 trials. The authors also thank Katarina Cepo, Søren Tan Camer, Jesper Haaning, Christian Hollensen, Ulla Ivens, Matilde Thye Kveiborg, and Thomas Porstmann from Novo Nordisk for their review and input to the manuscript.
Medical writing support was provided by Physicians World Europe GmbH, Mannheim, Germany, and was financially supported by Novo Nordisk A/S, Denmark.
Physicians World Europe provided medical writing support for the development of the first draft of the manuscript following an author meeting in which the authors assessed the available data and provided specific feedback as to the content of the initial draft.
Authorship
Contribution: All authors were investigators in either the explorer4 or explorer5 trials, recruited patients into the studies, analyzed and interpreted the data, contributed to subsequent writing and review of the manuscript, and approved the final version for submission.
Conflict-of-interest disclosure: A.D.S. serves as principal investigator on 3 Novo Nordisk–sponsored research studies. J.A. has received honoraria and consulting fees from Bayer, CSL Behring, Novo Nordisk, Octapharma, Roche, Sobi, Spark, Takeda, and uniQure. G.B. has received grants and personal fees for lectures and consultancy from Bayer, Boehringer Ingelheim, CSL Behring, Novo Nordisk, Pfizer, Shire, and Sobi. G.C. participated in a uniQure advisory board meeting; received fees to act as a speaker from, or to participate in advisory board meetings for, Ablynx, Bayer, CSL Behring, Kedrion, Novo Nordisk, Shire/Takeda, Sobi, Roche, and Werfen; and received research grants from CSL Behring, Pfizer, and Sobi. P.C. has received honoraria from Baxalta/Shire, Biogen Idec, CSL Behring, Novo Nordisk, Pfizer, Roche and Sobi; has served on advisory boards for Bayer, Baxalta/Shire, Biogen Idec, CSL Behring, Chugai, Freeline, Novo Nordisk, Pfizer, Roche, and Sobi; and has received research funding from Bayer, CSL Behring, Novo Nordisk, Pfizer, and Sobi. H.E. has received fees to act as a speaker or consultant from, or to participate in advisory board meetings for, Bayer, CSL Behring, Novo Nordisk, Shire/Takeda, Sobi, and Roche, and has received research grants from Bayer Vital, CSL Behring, and Pfizer. V.J.-Y. has received reimbursement for attending symposia/congresses and/or honoraria for speaking and/or consulting and/or funds for research from Bayer, CSL Behring, Grifols, Novo Nordisk, Octapharma, Pfizer, Roche, Sobi, and Shire. K.K. has participated in advisory board meetings for Bayer, Novo Nordisk, Takeda, Pfizer, and Roche. T.M. has received honoraria from Bayer, Bioverative, Chugai, CSL, KM Biologics, Novo Nordisk, and Shire, and research support from Bayer and Bioverative. L.H.P. has received funding for attending congresses and meetings from Bayer, Novo Nordisk, Pfizer, and Sobi. A.P.W. has participated in advisory board meetings for Biomarin, Novo Nordisk, Octapharma, Shire, and uniQure. G.Y. has received honoraria and consulting fees from Bioverativ/Sanofi, CSL Behring, Genentech/Roche, Grifols, Kedrion, Novo Nordisk, Spark, Takeda, and uniQure. S.Z.-S. has received reimbursement for attending symposia and congresses, and honoraria payment for speaking, from Biogen, Novo Nordisk, Sobi, and Roche. J.O. has received reimbursement for attending symposia/congresses and/or honoraria for speaking and/or consulting and/or funds for research from Bayer, Biogen Idec, Biotest, Chugai, CSL Behring, Grifols, Novo Nordisk, Octapharma, Pfizer, Roche, Taked, and Swedish Orphan Biovitrum. P.A. declares no competing financial interests.
Correspondence: Amy D. Shapiro, Indiana Hemophilia and Thrombosis Center, 8326 Naab Rd, Indianapolis, IN 46260; e-mail: ashapiro@ihtc.org.


