

In this issue of Blood, identified a causal link between a platelet signaling defect and bleeding in patients with Noonan syndrome (NS) who have activating mutations of Src homology 2 (SH2) domain-containing phosphatase 2 (SHP2).1
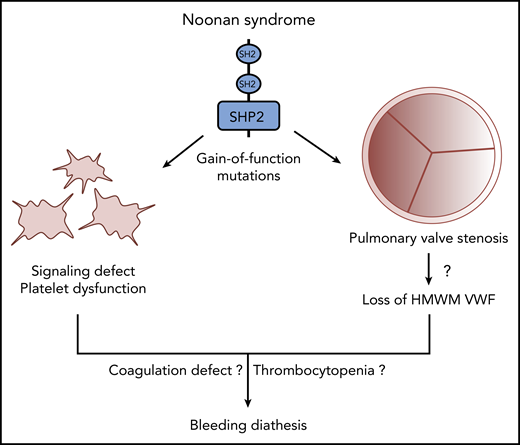
Multiple mechanisms likely contribute to disordered bleeding in patients with NS caused by gain-of-function mutations of SHP2.
Multiple mechanisms likely contribute to disordered bleeding in patients with NS caused by gain-of-function mutations of SHP2.
NS is a developmental disorder affecting 1 in every 1000 to 2500 people.2 This clinically and genetically heterogeneous syndrome is commonly characterized by defects in the RAS/MAPK cell signaling pathway. Although multiple gene mutations can cause NS, about half the cases are caused by mutations in the PTPN11 gene. Affected individuals most often present with distinctive facial features, short stature, chest deformity, and a wide spectrum of congenital heart diseases.
Disordered bleeding has been reported for 30% to 65% of individuals with NS and has consistently been associated with mutations in the PTPN11 gene. Bleeding is often mild with symptoms such as excessive bruising, epistaxis, or menorrhagia, but it can become severe after surgical procedures. Defects of 1 or several coagulation factors or thrombocytopenia have been described but without any consistent association with bleeding episodes.
It is noteworthy that the study by Bellio et al is the first to describe a genetic cause of thrombopathy resulting from a platelet signaling defect linked to disordered bleeding. So far, only a few inherited platelet disorders have been related to a platelet signaling defect such as autosomal recessive gene mutations in PLA2G4A (Ghosal syndrome), TBXSA1, and GNAS.3 However, whether these specific platelet defects are responsible for bleeding remains unknown.
The study by Bellio et al provides the first mechanistic explanation for bleeding in patients with PTPN11 mutations. The authors used genetically engineered mouse models and blood samples from NS patients to demonstrate that mutations of this gene can cause a platelet signaling defect. The protein encoded by PTPN11 is the ubiquitously expressed classical nonreceptor protein tyrosine phosphatase (PTP) SHP2.4 This protein has 2 tandem N-terminal SH2 domains, a catalytic PTP domain, a C-terminal tail with 2 tyrosine phosphorylation sites (Y542 and Y580), and a proline-rich region. At resting state, SH2 domain blocks access to SHP2 substrates by binding to its active site pocket. Upon ligand-receptor interaction, the N-SH2 domain preferentially binds to tyrosine-phosphorylated proteins such as receptor tyrosine kinase or scaffold proteins to open up the phosphatase active site, resulting in catalytic activation of SHP2. Mutations causing NS result in SHP2 gain-of-function by destabilizing the catalytically inactive conformation of the protein, which prolongs ligand-dependent signal transduction. In addition to NS, loss-of-function mutations of SHP2 can cause a rare NS-related syndrome, NS with multiple lentigines (NSML). Intriguingly, patients with NSML can also present with disordered bleeding.
Previous studies in mice with megakaryocyte- and platelet-specific deficiencies indicate that SHP2 contributes to thrombus stabilization under high shear stress without dramatically interfering with hemostasis.5 Indeed, platelet aggregation induced by collagen, adenosine diphosphate, or thrombin occurred normally in the absence of SHP2. In sharp contrast to these findings, Bellio et al showed a proximal collagen receptor signaling defect in mice heterozygous for the gain-of-function D61G mutation in SHP2. Ex vivo and in vivo thrombus formation were also impaired, and tail bleeding time was prolonged in most animals, proving the defective primary hemostasis. Hence, these data nicely recapitulated the bleeding diathesis of NS patients with gain-of-function mutations of SHP2. These results were validated with blood samples from NS patients who bear different gain-of-function mutations of SHP2. Despite variable Tosetto bleeding scores ranging from normal to high, platelets from all patients displayed defective aggregation in response to low concentrations of collagen as well as impaired thrombus formation on a collagen-coated surface under flow. Conversely, platelets from mice expressing a loss-of-function SHP2 mutant showed enhanced response to low concentrations of collagen, which further confirmed the role of SHP2 in regulating collagen receptor signaling. Thrombus formation was also increased under high shear stress. Ferric chloride–induced occlusive thrombosis and bleeding time were normal, in agreement with the existence of a mild shear-dependent prothrombotic tendency associated with loss-of-function mutations of SHP2. Accordingly, blood samples from 3 NSML patients depicted enhanced thrombus growth on a collagen surface under high shear stress. Platelet aggregation induced by collagen occurred normally, which further excluded major platelet hyperreactivity under low nonpathological shear stress.
Another prominent feature of NS caused by mutations in PTPN11 is pulmonary valve stenosis (PVS).2,6 In NS, because of tissue overgrowth, pulmonary valve leaflets are thickened and often dysplastic, leading to impaired valve mobility and opening, which hinders blood flow from the right ventricle to the lungs. This condition can result in obstruction of the right ventricular outflow tract, which necessitates surgical or transcatheter pulmonary valve replacement (PVR).7 In PVS, valve stenosis increases the systolic pressure gradient, which can reach peak values >64 mmHg. In severe aortic stenosis (AS), it has been reported that a high pressure gradient and the resulting high shear stress induce cleavage of von Willebrand factor (VWF) by the metalloproteinase ADAMTS-13, leading to secondary loss of high-molecular-weight multimer (HMWM) VWF.8 Nevertheless, the association between the loss of HMWMs and AS bleeding remains uncertain. But because cleaved VWF interacts less efficiently with platelets and collagen, this phenomenon could at least partially explain the occurrence of acquired von Willebrand disease and Heyde’s syndrome in AS. Interestingly, even though the pressure gradients are lower in PVS than in AS, a small study conducted with 15 NS patients suggested a relationship between PVS severity, loss of HMWMs, and bleeding diathesis in some of the patients.9 Thus, on the basis of that study and the work of Bellio et al, we can assume that multiple factors are likely to contribute to bleeding in NS patients, including platelet signaling defects, coagulation impairments, severity of PVS, and subsequent loss of HMWMs (see figure). Hence, thorough characterization of platelet function (eg, platelet aggregometry and shear-dependent PFA-200 analysis), coagulation, and heart valve defects should be part of NS clinical management to anticipate any procedural risk of major bleeding, particularly in patients undergoing PVR. Finally, further studies should investigate the association between PVS severity, platelet dysfunction, and bleeding not only in NS but also in PVS from other etiologies.6
Conflict-of-interest disclosure: C.O. is Research Director at National Funds for Scientific Research, Belgium (FRS-FNRS). P.L. declares no competing financial interests.

