

In this issue of Blood, have expanded the territory in our search for pathogenetic factors in cancer-associated venous thromboembolism (VTE).1 Such factors, which could potentially be used as biomarkers or therapeutic targets, are usually elusive when chased and ephemeral when caught. Time has not been kind to many of the previously identified factors, from D-dimers to tissue factor (TF)–bearing microparticles,2 and it will be interesting to see whether the hypothesis that the vascular endothelial cell (EC) aryl hydrocarbon receptor (AHR) contributes to the development of cancer-associated venous thrombosis withstands the test of time.
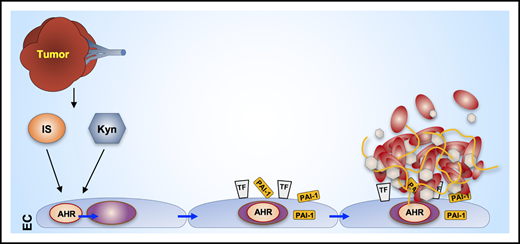
Tumor-associated production of indoxyl sulfate (IS) and kynurenine (Kyn) bind to the EC AHR, thereby stimulating EC production of prothrombotic TF and PAI-1, leading to fibrin thrombus formation (see Figure 9 in the article by Belghasem et al that begins on page 2399).
Tumor-associated production of indoxyl sulfate (IS) and kynurenine (Kyn) bind to the EC AHR, thereby stimulating EC production of prothrombotic TF and PAI-1, leading to fibrin thrombus formation (see Figure 9 in the article by Belghasem et al that begins on page 2399).
The AHR is member of the basic helix-loop-helix/Per-Arnt-Sim superfamily of transcription factors. About 250 million years ago, the metazoan AHR began to evolve. Its evolution was driven by adaptation to environmental toxins. In humans, it was identified because of its function to bind to toxins such as 2,3,7,8-tetrachlorodibenzo-p-dioxin, translocate to the nucleus, and activate genes of metabolic systems (principally cytochrome P4501A1) that detoxify the cell.3 Its conservation in metazoans, from clams to humans, indicates that it serves an important physiological function, but to date, the hierarchy of its physiological effects remains unclear. Deletion of the AHR in mice results in many phenotypic changes, but notably, no systemic vascular abnormalities.4
Accumulating experimental evidence has identified numerous endogenous ligands regulating the AHR in a variety of physiological and pathological conditions, including atherothrombosis5 and cancer.6 Two of the endogenous ligands are kynurenine, produced from tryptophan by the cellular enzymes indoleamine 2,3-dioxygenase and tryptophan 2,3-dioxygenase and indoles produced from gut microbiome metabolism of tryptophan.7 Data presented by Belghasem et al demonstrate that human colorectal cancer xenografted into athymic nude mice leads to increased plasma concentrations of kynurenine (probably synthesized by the tumor) and indoxyl sulfate (of presumed microbial origin) and that xenografted animals develop more thrombi after inferior vena cava (IVC) ligation. The phenomena are coupled mechanistically by data showing that plasma from tumor-bearing animals activates an AHR-specific EC reporter and stimulates EC TF activity and plasminogen activator inhibitor 1 (PAI-1) protein expression, both of which are inhibited by an AHR antagonist. Direct inhibitable effects of kynurenine on TF activity, PAI-1 expression and clot weight, are shown in mice without xenografts. Finally, tumor-associated activation of endothelial AHR, AHR-dependent expression of endothelial TF and PAI-1, and AHR-dependent thrombus formation are shown to occur within the IVC.
The pathophysiological model presented by the investigators (see figure) also provides a map of the information gaps that one must fill before AHR-mediated thrombosis is proven relevant and actionable. Do other cancers have the same prothrombotic effects and is it because of their synthesis of kynurenine? What happens with an intact immune system? Is indoxyl sulfate gut derived? And what are mechanisms of tumor-gut interactions that control indole synthesis? How significant is the AHR pathway as a general prothrombotic signal? Can it cause thrombosis separately from major vessel trauma in the mouse model? Is it relevant in humans? Could it be involved in idiopathic thrombosis? What other prothrombotic signals activate the AHR response? And what other thrombosis-regulating genes are activated by the AHR? Only after these and derivative questions are answered will we begin to understand whether the findings in the Belghasem article have implications for human disease. As stated by the authors, there is a “staggering variability in patient- and cancer-related VTE risk.” It is up to them and the scientific community to prove that the AHR is a general and significant element contributing to the pathogenesis of cancer-associated VTE.
Conflict-of-interest disclosure: The author declares no competing financial interests.

