Key Points
CD19 CAR-T cell immunotherapy results in high CR rates in FL with or without transformation.
Sustained remissions were observed in all patients with FL without transformation achieving CR.

Abstract
Patients with follicular lymphoma (FL) with early relapse after initial chemoimmunotherapy, refractory disease, or histologic transformation (tFL) have limited progression-free and overall survival. We report efficacy and long-term follow-up of 21 patients with relapsed/refractory (R/R) FL (n = 8) and tFL (n = 13) treated on a phase 1/2 clinical trial with cyclophosphamide and fludarabine lymphodepletion followed by infusion of 2 × 106 CD19-directed chimeric antigen receptor–modified T (CAR-T) cells per kilogram. The complete remission (CR) rates by the Lugano criteria were 88% and 46% for patients with FL and tFL, respectively. All patients with FL who achieved CR remained in remission at a median follow-up of 24 months. The median duration of response for patients with tFL was 10.2 months at a median follow-up of 38 months. Cytokine release syndrome occurred in 50% and 39%, and neurotoxicity in 50% and 23% of patients with FL and tFL, respectively, with no severe adverse events (grade ≥3). No significant differences in CAR-T cell in vivo expansion/persistence were observed between FL and tFL patients. CD19 CAR-T cell immunotherapy is highly effective in adults with clinically aggressive R/R FL with or without transformation, with durable remission in a high proportion of FL patients. This trial was registered at clinicaltrials.gov as #NCT01865617.
Introduction
Treatment of relapsed/refractory (R/R) large B-cell non-Hodgkin lymphoma (NHL) patients with the CD19-directed chimeric antigen receptor–modified T (CAR-T) cell products, axicabtagene ciloleucel and tisagenlecleucel, results in overall response rates (ORRs) of 82% and 52%, respectively, with most relapses occurring in the first 12 months after infusion.1,2 Follicular lymphoma (FL) is the second most frequent NHL subtype. Patients who relapse within 2 years after initial chemoimmunotherapy have limited survival (5-year overall survival [OS], 50%),3 as do those who fail multiple regimens (5-year progression-free survival [PFS], 23%)4 or develop histologic transformation to large-cell NHL (median PFS 1 year).5 Initial responses after CD19 CAR-T cell immunotherapy have been reported in patients with R/R FL and were often durable.6-8 Here, we report the incidence and durability of responses in patients with FL without transformation or with histologic transformation (tFL) after treatment with CD19 CAR-T cells of defined CD4+:CD8+ CAR-T cell composition.
Study design
We conducted a phase 1/2 clinical trial (#NCT01865617) of CD19 CAR-T cell immunotherapy in adults with R/R CD19+ B-cell malignancies.9-11 The study was conducted according to the Declaration of Helsinki with informed consent and approval by the Institutional Review Board. Here, we report all patients with FL and tFL who received lymphodepletion with a cyclophosphamide and fludarabine–containing regimen followed by 2 × 106 CD19 CAR-T cells per kilogram, formulated at a 1:1 CD4+:CD8+ CAR-T cell ratio. Manufacturing of CD19 CAR-T cells with 4-1BB costimulation has been described.9,10 Bridging antitumor therapy was permitted after leukapheresis. Patients were monitored for response and toxicities, as previously described.10,12 Best responses were reported according to the Lugano criteria.13 Kaplan-Meier (KM) analyses were used to estimate PFS, duration of response, and OS. Follow-up time was estimated by a reverse KM estimator.14 Fisher’s exact and Wilcoxon rank-sum tests were used to compare categorical and noncategorical variables, respectively. Data were analyzed using R version 3.4.1 and RStudio version 1.0.153.
Results and discussion
Twenty-one patients (median age, 56 years) who received a cyclophosphamide and fludarabine lymphodepletion regimen and 2 × 106 CD19 CAR-T cells per kilogram are included in this study (Table 1). Eight patients (38%) had FL, and 13 (62%) had tFL.
The FL patients had received a median of 4 prior treatment regimens (range, 2-7); all had failed chemoimmunotherapy, including an anti-CD20 antibody and alkylating agents. Seven of 8 patients had failed prior anthracycline exposure; 75% had progressive disease after the last therapy, and 50% (n = 4) had failed prior autologous (n = 3) or allogeneic (n = 1) hematopoietic cell transplantation (HCT). The 4 FL patients who had not failed HCT were not considered suitable candidates for HCT due to refractory disease. One FL patient received bridging chemotherapy to control progressive disease between leukapheresis and lymphodepletion. Before lymphodepletion, 75% of FL patients had stage III or IV disease; 62% had extranodal disease, and 75% had intermediate or high FLIPI score.15 The median tumor burden, estimated by the sum of the product of the perpendicular diameters of up to 6 index lesions, was 2995 mm2. Our patients had high-risk characteristics comparable to FL patients treated in another trial of CD19 CAR-T cell therapy.8
Twelve of the 13 patients with tFL had documented diffuse large B-cell lymphoma (DLBCL) transformation at the last biopsy. One patient with aggressive, bulky, and intensely FDG-avid lymphadenopathy had focal grade 3B disease on the last biopsy and was included in the tFL group. Four patients had confirmed high-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements (double-/triple-hit lymphoma). The median time from first histologic documentation of transformation to prelymphodepletion evaluation was 17.9 months (range, 7-83). The tFL patients had received a median of 5 prior treatment regimens (range, 2-8), and 62% (n = 8) had failed prior autologous (n = 6), allogeneic (n = 1), or both autologous and allogeneic (n = 1) HCT. Between leukapheresis and lymphodepletion, 4 of 13 tFL patients (31%) required systemic bridging therapy to control disease progression. Before lymphodepletion, all patients had stage III or IV disease; 92% had extranodal disease, and 92% had intermediate or high FLIPI score. The median tumor sum of the product of the perpendicular diameters was 4695 mm2. Patients with FL and tFL had comparable baseline and treatment characteristics (Table 1); however, more tFL patients had elevated LDH (FL vs tFL, 13% vs 69%, P = .02), and fewer had bone marrow involvement (50% vs 15%, P = .15).
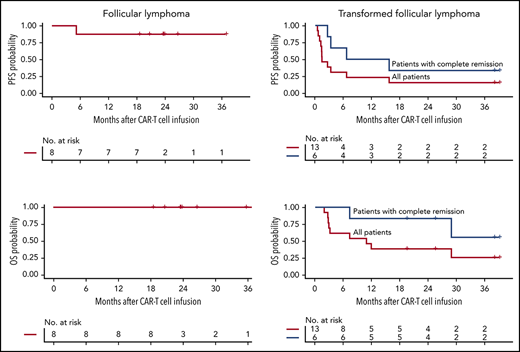
Seven of 8 patients with FL achieved complete remission (CR; 88%) after CAR-T cells. The median time to CR was 29 days (range, 27-42), and all who achieved CR remained in remission (median follow-up, 24 months, range, 5-37). One patient received additional therapy (allogeneic HCT) while still in CR. One patient with stable disease at first restaging received radiation 2.3 months after CAR-T cells and has not progressed 36 months after CAR-T cell infusion. These data demonstrate a high rate of durable CR in high-risk FL patients treated with CD19 CAR-T cells, comparable to that reported in another study.8 The CD19 CAR-T cell products, axicabtagene ciloleucel and tisagenlecleucel, are also being studied in FL patients in ongoing clinical trials.16,17
For the 13 patients with tFL, the best ORR without additional therapy after CAR-T cells was 46%, with all responding patients achieving CR. Three of 4 patients with tFL with double-/triple-hit lymphoma had progressive disease. For those who achieved CR, at a median follow-up of 38 months (range, 3-39), the median PFS was 11.2 months (95% confidence interval [CI], 3.3-NR [not reached]). For all patients with tFL, the median duration of response and PFS were 10.2 months (95% CI, 2.3-NR) and 1.4 months (95% CI, 1.2-NR), respectively (Figure 1). No relapses had occurred after 15.6 months, with durable remissions observed for up to 39 months after CAR-T cell infusion.

PFS and OS in patients with FL after CD19 CAR-T cell immunotherapy. KM estimates of PFS (A-B) and OS (C-D) in patients with FL (A-C) and transformed FL (B-D) who achieved CR (blue) and in all patients (red). The numbers of patients at risk at 6-month intervals are indicated.
PFS and OS in patients with FL after CD19 CAR-T cell immunotherapy. KM estimates of PFS (A-B) and OS (C-D) in patients with FL (A-C) and transformed FL (B-D) who achieved CR (blue) and in all patients (red). The numbers of patients at risk at 6-month intervals are indicated.
CD19 CAR-T cell immunotherapy was well tolerated in most patients. No significant differences were observed in the incidence or severity of cytokine release syndrome (grade ≥1; FL vs tFL, 50% vs 39%, P = .35), or neurotoxicity (grade ≥1; FL vs tFL, 50% vs 23%, P = .67) between FL and tFL patients. Severe (grade ≥3) cytokine release syndrome and neurotoxicity were not observed. Although CR and PFS rates differed between FL and tFL patients, the peak CAR-T cell counts and the area under the curve until day 28 (AUC 0-28) by quantitative polymerase chain reaction in these groups were similar. The duration of CAR-T cell detection by quantitative polymerase chain reaction was also similar (FL, median 4.9 months, range, 0.9-24.0; tFL, median 3.0 months, range 0.5-24.5; P = .33), as were pre-lymphodepletion, day 0, peak, and AUC 0-28 serum interferon-γ, interleukin-2Rα (IL-2Rα), IL-5, IL-6, IL-7, IL-10, IL-15, IL-18, IL-22, monocyte chemoattractant protein-1, macrophage inflammatory protein-1β, soluble Fas, soluble IL-6R, transforming growth factor β-1, T-cell immunoglobulin, mucin domain-3, tumor necrosis factor (TNF)-α, TNF receptor p55, and TNF receptor p75 concentrations.
The serum IL-8 concentration on day 0 before CAR-T cell infusion was higher in tFL compared with FL patients (median, 9.6 vs 2.5 pg/mL, P = .01). IL-8 has been described to mediate the recruitment of tumor-associated neutrophils and promote DLBCL progression18 and can contribute to local immune suppression.19 Patients with tFL had also higher pretreatment LDH, which could reflect more aggressive disease and a more immunosuppressive tumor microenvironment.20-23 We have already reported in aggressive NHL patients the association between high LDH concentrations and poor outcomes after CD19 CAR-T cell immunotherapy.12 Although these data raise the possibility that differences in the tumor microenvironment may in part contribute to differences in outcomes after CAR-T cell immunotherapy in FL and tFL patients, additional studies are required.
CD19 CAR-T cell immunotherapy was well tolerated and resulted in a high rate of CR (88%) in patients with clinically aggressive R/R FL, and all patients remained in CR with sustained remissions lasting up to 3 years after CAR-T cell infusion. These data indicate that further development of CD19 CAR-T cell immunotherapy for patients with high-risk FL is warranted. The CR rate in tFL (46%) was lower than in FL, but compared favorably with the SCHOLAR-1 study (ORR, 26%; CR rate, 7%)24 and similarly to reported responses of patients with de novo DLBCL after CD19 CAR-T cell immunotherapy.1,2,8
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the staff of the Fred Hutchinson Cancer Research Center (FHCRC) Cell Processing Facility, Seattle Cancer Care Alliance Cell Therapy Laboratory, the FHCRC Integrated Immunotherapy Research Center, and the Seattle Cancer Care Alliance Bezos Family Immunotherapy Clinic.
This work was supported by National Institutes of Health (NIH), National Cancer Institute grants R01 CA136551 and P30 CA15704, NIH National Institute of Diabetes and Digestive and Kidney Diseases grant P30 DK56465, NIH National Heart, Lung, and Blood Institute–funded National Gene Vector Biorepository at Indiana University (contract 75N92019D00018), Life Science Discovery Fund, the Bezos family, the University of British Columbia Clinician Investigator Program, FHCRC Immunotherapy Integrated Research Center, and Juno Therapeutics/Celgene, Inc.
Authorship
Contributions: A.V.H., J.G., and K.A.H. collected and analyzed research data; B.S.P., R.M.H., A.V., and R.N.S. performed experiments; J.M.V. and Q.W. performed statistical analyses; A.V.H. and C.J.T. wrote and edited the manuscript; and all authors reviewed and edited the final version of the manuscript.
Conflict-of-interest disclosure: K.A.H. has served on advisory boards for Celgene; S.R.R. holds equity, has served as an advisor, and has patents licensed to Juno Therapeutics, a Celgene/Bristol-Myers Squibb company; is a founder of Lyell Immunopharma; and has served on advisory boards for Adaptive Biotechnologies and Nohla; D.G.M. has received research funding from Kite Pharma, a Gilead Company, Juno Therapeutics, a Celgene/Bristol-Myers Squibb company, and Celgene; has served on advisory boards for Kite Pharma, Gilead, Genentech, Novartis, and Eureka Therapeutics; C.J.T. receives research funding from Juno Therapeutics, a Celgene/Bristol-Myers Squibb company, and Nektar Therapeutics; has patents licensed to Juno Therapeutics, a Celgene/Bristol-Myers Squibb company; has served on advisory boards and has equity ownership in Caribou Biosciences, Eureka Therapeutics, and Precision Biosciences; and has served on advisory boards for Aptevo, Juno Therapeutics, a Celgene/Bristol-Myers Squibb company, Kite Pharma, a Gilead Company, Humanigen, Nektar Therapeutics, Allogene, and Novartis. The remaining authors declare no competing financial interests.
Correspondence: Alexandre V. Hirayama, Clinical Research Division, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave N, Seattle, WA 98109; e-mail: ahirayama@fredhutch.org.