

The initial stages of thymopoiesis are characterized by the induction of T-lineage genes (specification) and the repression of alternative lineage genes (commitment). Multilineage (Thy1 and Thy2) and committed (Thy3) populations representing successive differentiation stages have been defined within CD34+ progenitor cells in the human thymus by the expression of CD7 and CD1a. However, due to intra-population heterogeneity and unstudied transitional states between populations, our understanding of the transcriptional programs that launch human thymopoiesis remains incomplete. Also, the interpretation of species specific gene expression profiles seen in these populations is confounded by species related differences in progenitor immunophenotypes. To our knowledge, whole transcriptome profiles of human thymic CD34+ progenitors at single cell resolution have not been reported.
To resolve the transcriptional landscape of the initial stages of thymopoiesis, we performed single cell RNA-Seq (sRNA-Seq) of FACS sorted unfractionated human thymic CD34+CD4-CD8- cells from 3 donors using 10X (1 thymus, 5000 cells), Indrop (1 thymus, 2000 cells), or BD genomics (1 thymus, 192 index sorted cells) sequencing Averaged sRNA-Seq expression profiles recapitulated previous bulk RNASeq data validating our sRNA-Seq pipeline. Transcriptomes of index sorted cells mapped appropriately to those predicted to be Thy1,Thy2, or Thy3 cells from CD34, CD7, and CD1a RNA levels in the 10x and indrops data indicating the validity of identifying Thy1-3 cells by RNA levels.
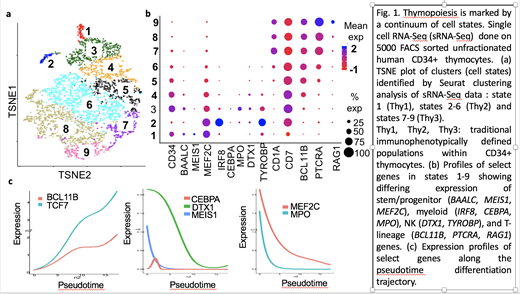
Seurat clustering showed that CD34+ thymocytes consist of a continuum of cell states rather than discrete populations. Nine cell states marked by gradual changes in CD2, CD44, CD7, CD1A, and CD34 RNA levels were seen: state 1 (Thy1), states 2-6 (Thy2), and states 7-9 (Thy3). Thy2 and Thy3 cells spanned several previously undescribed states with distinct expression profiles. Single cells co-expressing stem and T-lineage genes were seen in state 1 indicating an early onset of T-lineage priming that occurs prior to downregulation of stem cell genes. Within Thy2 cells, high expression of stem and alternative lineage genes was mostly restricted to states 2 and 3, which represent novel Thy2 subpopulations. State 2 cells co-expressed T-cell and high levels of innate immune genes, particularly IRF8. State 3 cells showed low CD2 expression and co-expressed T-cell and stem, myeloid, B-cell, and/ or NK genes. These results indicate concomitant priming of multiple lineage transcriptional programs rather than just the presence of unilineage gene program expressing subpopulations in the earliest thymic progenitor cells. In preliminary experiments, FACS sorted CD2 low Thy2 cells generated T-cells as well as a higher output of alternative lineage cells than CD2 high Thy2 cells.
States 7-9 showed committed profiles with upregulated T-cell genes and silenced stem and alternative lineage genes. While state 9 (late Thy3) showed high RAG1 expression and cell cycle arrest, most Thy3 cells (states 7 and 8) were cycling indicating that commitment, which precedes T-cell receptor rearrangement, is not associated with proliferation arrest. Monocle pseudotime analysis finely resolved expression profiles along the differentiation trajectory within Thy1-3 populations: e.g. genes repressed earlier (e.g. MEIS1) vs later (e.g. DTX1). BCL11B, a key T-cell commitment gene was one of the earliest transcription factors (TF) to be induced during human thymopoiesis, and upregulation of TCF7 and BCL11B occurred concurrently. In contrast, in mice induction of Bcl11b occurs later after Tcf7 is fully upregulated.
Thus, the initial stages of thymopoiesis are characterized by a continuum of multilineage transcriptional priming followed by a gradual transition to a T-lineage restricted gene program, a process similar to that recently described for lineage commitment of bone marrow hematopoietic progenitors but not previously reported in the thymus. SRNA-Seq yielded expression profiles along numerous data-points on the differentiation trajectory within heterogenous populations, providing high transcriptional resolution not possible with bulk data. We found differences in key TF expression profiles that could imply species specific regulatory mechanisms. Our data are a resource for elucidating transcriptional networks in T-cell development and T-cell leukemias.
No relevant conflicts of interest to declare.

