

Background:
Therapy for chronic anemias is limited to RBC transfusions and Erythropoiesis Stimulating Agents (ESA) which often work on only transiently or not at all. New approaches to treat chronic anemia are needed but development has been limited by our incomplete understanding of erythropoiesis, most of which relates to the terminal maturation of erythroid precursors. Erythropoietin (Epo) acts during a very narrow window of erythropoiesis, well after progenitor commitment to an exclusively erythroid fate. It is not known if the final steps of RBC maturation are coupled to the earlier stages of hematopoietic stem and progenitor cell (HSPC) differentiation; a process that begins almost three weeks earlier when an HSC starts its march towards committed RBC precursors via a series of branching cell fate decisions.We searched for independent control and compartmentalization of erythropoiesis that could couple early hematopoiesis to terminal erythroid commitment and maturation.
Results:
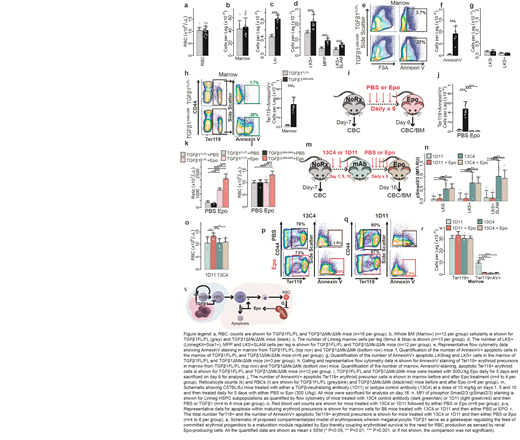
We deleted TGFβ1 in megakaryocytes (TGFβ1ΔMk/ΔMk) and found that peripheral blood counts were normal in TGFβ1ΔMk/ΔMkmice compared to TGFβ1FL/FLcontrols despite the pool of primitive hematopoietic cells being expanded (Fig. 1a). Similarly total bone marrow cellularity was normal in TGFβ1ΔMk/ΔMkmice (Fig. 1b). Excess HSCs in TGFβ1ΔMk/ΔMkmice appeared capable of robust differentiation because the number of immature lineage-negative (Linneg) hematopoietic progenitor cells was increased in the marrows of TGFβ1ΔMk/ΔMkmice (Fig. 1c). Thus, it remained unexplained why the expanded number of HSPCs (Fig. 1d) do not increase blood counts and marrow cellularity.
We hypothesized that the excess progenitors observed in the TGFβ1ΔMk/ΔMkmice failed to increase blood counts because their progeny were unneeded, and inadequately supported by homeostatic levels of late-acting cytokines. Indeed, bone marrow apoptosis was increased in the TGFβ1ΔMk/ΔMkmice compared to controls, as reported by AnnexinV (AV) binding (Fig. 1e-f). Apoptosis of lineage-marker negative (Linneg), Kit+Sca1neg(LKSneg) HPCs and LKS+HSPCs was rare in both TGFβ1ΔMk/ΔMkmice and littermate controls (Fig. 1g). These results suggest that excess, hematopoietic precursors present in the TGFβ1ΔMk/ΔMkmice are pruned by apoptosis during hematopoietic differentiation.
We found 10-fold apoptosis in TGFβ1ΔMk/ΔMkprecursors populations BM (Fig. 1h). Epo levels were normal in the serum of these mice, we reasoned that the excess, unneeded cells were not supported physiologic Epo levels. To test this, we treated mice with exogenous Epo. Indeed, we found that the excess erythroid apoptosis could be rescued by administration of very low doses of Epo (300U/kg)(Fig. 1i-j). Whereas TGFβ1Flox/Floxmice showed minimal reticulocytosis and no change in blood counts, TGFβ1ΔMk/ΔMkmice responded with reticulocytosis and erythrocytosis within 6 days (Fig. 1k-l). In contrast, treatment of mice with TGFβ1 worsened the erythroid apoptosis observed in TGFβ1ΔMk/ΔMkmice and caused mild anemia. These results suggest that erythropoiesis is subject to modular regulation with megakaryocytic TGFβ1 constraining the pool of erythroid committed progenitors that are then licensed to mature via Epo signaling. We thought that blockade of TGFβ signaling could phenocopy these effects by inducing overproduction of erythroid committed precursors. To test this, we pre-treated B6 mice with a TGFβ1 neutralizing antibody (1D11) or non-targeting, isotype control antibody (13C4) and then either PBS or low-dose Epo (Fig. 1m). TGFβ neutralization by 1D11 reduced pSmad2/3 MFI in HSPCs in wild-type mice whereas the 13C4 control had no effect, demonstrating on-target activity (Fig. 1n). Low-dose Epo triggered a brisk erythropoietic response in mice treated with 1D11 but not those treated with the 13C4 control (Fig. 1o). Exogenous Epo rescued the erythroid precursor dropout observed in B6 mice treated with 1D11 but did not affect the low apoptosis observed in mice treated with the 13C4 control (Fig. 1p-r). Therefore, the boundary of megakaryocytic TGFβ1 activity is compartmentalized within the marrow with predominant effects on immature HSPCs while excluding their progeny (Fig. 4s).
Conclusion:
This work also promises new therapies for chronic anemias by combining TGFβ inhibitors to increase the outflow of immature progenitors with ESAs to support erythroid maturation.
No relevant conflicts of interest to declare.

