

Combination antiretroviral therapies (cART) have markedly reduced mortality in HIV infection. However, cardiovascular disease (CVD), including heart failure linked to fibrosis, remains a major cause of morbidity and mortality in HIV/cART patients. The magnitude of this risk increases with use of certain protease inhibitors (PI), but the underlying mechanism remains unclear. We showed that the PI ritonavir leads to increased plasma levels of the pro-fibrotic cytokine TGF-β1, cardiac dysfunction, and pathologic cardiac fibrosis in wild-type (wt) C57BL/6 mice. Mice with targeted depletion of platelet TGF-β1 had reduced cardiac fibrosis and partially preserved cardiac function following ritonavir exposure (Laurence, et al. PLoS One 2017;12:e0187185). Several groups have examined the effects of a variety of cART agents on agonist-induced platelet aggregation, but correlations with clinical CVD are weak. Since platelets are a rich source of TGF-β1, we hypothesized that ritonavir and other PIs linked clinically to an increased CVD risk directly activate platelets to release TGF-β1 and activate latent (L)TGF-β1 to initiate signaling for organ fibrosis.
We examined the impact of clinically relevant doses of ritonavir, alone and in combination with two other contemporary PIs, atazanavir and darunavir, which are currently used along with low dose ritonavir in so-called PI-boosted cART regimens. We incubated human platelet-rich plasma and washed platelets with PIs alone or in combinations at various doses for 10 min at 37°C in a platelet aggregometer (BioData. Corp). Total and active TGF-β1 levels were measured by ELISA. For in vivo assessment, we treated wt mice with a low dose of ritonavir, as used in PI-boosted cART, and measured the levels of plasma TGF-β1 by ELISA, and TGF-β1 signaling in tissues by immunofluorescence imaging for pSmad2.
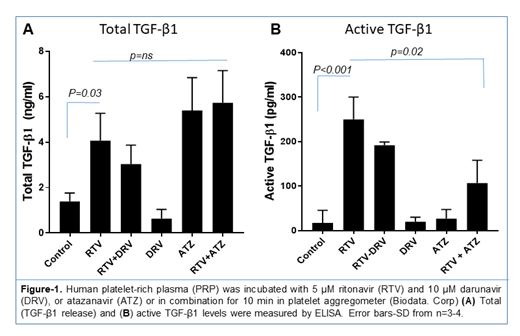
We found that ritonavir dose-dependently increased total TGF-β1 release from freshly-isolated platelet-rich plasma and washed human platelets. This release was blocked by ceefurin-1 and MK517, potent inhibitors of the ATP binding cassette transporter ABCC4. Darunavir alone did not cause release of TGF-β1, and did not alter significantly ritonavir-induced TGF-β1 release (Figure-1A). Atazanavir alone did induce release of TGF-β1 from platelets and did not affect the extent of such release induced by ritonavir (Figure-1A).
Since total TGF-β1 released from platelets must be activated in order to signal, we tested whether these PIs could activate LTGF-β1. Ritonavir alone, in low dose, activated TGF-β1 by 4-5-fold (Fig-1B). Darunavir alone did not activate LTGF-β1, and had only a minor effect on ritonavir-induced TGF-β1 activation (Fig-1B). In marked contrast, while atazanavir also did not activate LTGF-β1, it significantly inhibited ritonavir-induced LTGF-β1 activation (Fig-1B).
For in vivo assessment, wt mice were injected daily for 8 weeks with ritonavir, which dose-dependently increased plasma TGF-β1 levels (mean levels with vehicle 2.1 ng/ml; 6.4 ng/ml with 5 mg/kg ritonavir; 8.5 ng/ml with 10 mg/kg ritonavir). Increased TGF-β1 levels correlated with development of pathologic fibrosis and increased phosphorylated Smad signaling in hearts of ritonavir-treated vs. vehicle-treated mice.
Clinical correlations with these in vitro and in vivo mouse studies are important. The fact that ritonavir effected both release and activation of platelet TGF-β1 is consistent with its ability to induce cardiac fibrosis and dysfunction in mice, and its association with accelerated CVD in HIV-infected individuals. Our findings that low dose ritonavir in combination with darunavir induced release and activation of platelet TGF-β1, whereas atazanavir blocked TGF-β1 activation, are consistent with the strong association of ritonavir-boosted darunavir, but not ritonavir-boosted atazanavir, with CVD in the setting of HIV (Ryom, et al. Lancet-HIV 2018;5:e291-e300). Future work will examine the effects of other contemporary cART agents, including cobicistat, which is currently replacing ritonavir in many PI-boosted therapies and some integrase-boosted regimens, on TGF-β1 release and activation, for which correlations with clinical CVD are not yet available. Identification of the mechanism of pathologic fibrosis in the heart, and potentially other organs affected by certain cART regimens, such as the kidney, may suggest specific therapeutic interventions.
No relevant conflicts of interest to declare.

