

Background
Allogeneic hematopoietic cell transplantation (HCT) is indicated for several hematologic malignancies and nonmalignant disorders. Chronic graft-versus-host disease (cGVHD) develops in 30%-70% of long-term survivors of HCT and is a significant cause of nonrelapse mortality. Corticosteroids (CS) are the recommended first-line treatment for cGVHD; however, up to 60% of patients treated with CS require additional cGVHD therapy within 2 years. Furthermore, long-term exposure to CS is associated with a wide range of toxicities and increased risk of infection. Novel safe and effective therapies are needed to treat cGVHD and reduce CS dependence. Janus kinases (JAKs) mediate the signaling of proinflammatory cytokines involved in the pathogenesis of cGVHD. Itacitinib is a JAK-1 selective inhibitor that improved graft-versus-host disease (GVHD) and survival without affecting engraftment of donor leukocytes in murine models. In a phase 1 clinical study, itacitinib 200 or 300 mg once daily (QD) was well tolerated in patients with steroid-refractory or steroid-naive acute GVHD (aGVHD). Here we describe the trial design of the first clinical study evaluating the safety and efficacy of itacitinib for the treatment of cGVHD.
Study Design and Methods
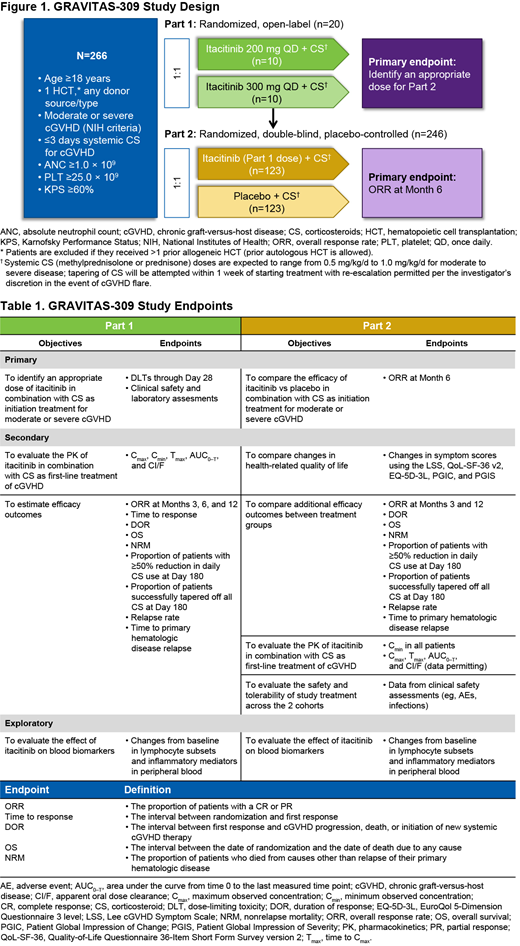
GRAVITAS-309 (NCT03584516) is a 2-part phase 3 study of itacitinib or placebo in combination with CS as initial treatment for cGVHD. Patients aged ≥18 years who underwent allogeneic HCT from any donor type and graft source, with evidence of myeloid and platelet engraftment, and who have active, moderate, or severe cGVHD per National Institutes of Health Consensus Criteria are eligible to participate. Patients are excluded if they received >1 prior HCT (except autologous), had >3 days of systemic CS treatment for cGVHD, received any other systemic treatment for cGVHD, have presence of active uncontrolled infection, or had treatment with a JAK inhibitor within 8 weeks of randomization (patients who previously received a JAK inhibitor for aGVHD are eligible only if they achieved a complete or partial response). Part 1 is a randomized, open-label study to determine optimal dosing of itacitinib in combination with CS (~20 patients randomized 1:1 to itacitinib 200 mg QD or 300 mg QD; Figure 1). The primary endpoints for Part 1 are dose-limiting toxicities (DLTs) through Day 28 as well as clinical safety and laboratory assessments. Main secondary endpoints include pharmacokinetic (PK) parameters and efficacy outcomes (Table 1). After 20 patients have completed 28 days of treatment, an external data monitoring committee will review the data and provide a recommendation for an appropriate dose for Part 2 based on emergent adverse events, clinical laboratory parameters, PK data, and DLTs. Part 2 is a randomized, double-blind, placebo-controlled study using the dose selected in Part 1 to assess the efficacy and safety of itacitinib plus CS vs placebo plus CS (~246 patients stratified by cGVHD severity and randomized 1:1; Figure 1). Patients randomized to the placebo group will be permitted to cross over to the itacitinib group after completion of the primary analysis. The primary endpoint is overall response rate (proportion of patients with complete or partial response) at 6 months. Secondary endpoints include changes in symptom scores using health-related quality-of-life measures, additional efficacy outcomes, PK parameters, and clinical safety assessments (Table 1). Exploratory biomarker analyses will evaluate the effects of JAK inhibition on circulating inflammatory cells (ie, T-cell subsets, B cells, natural killer cells, cytokines) and biomarkers of GVHD in peripheral blood after transplant. Patients will remain on study for a total of 37 months, which includes treatment period, safety follow-up, and posttreatment GVHD follow-up, unless confirmed GVHD progression, start of a new GVHD therapy, or relapse/recurrence of underlying hematologic disease occurs earlier. Efficacy analyses will be performed on the intent-to-treat population; safety analyses will be conducted on all randomized patients who receive ≥1 dose of study drug. The PK-evaluable population will include all patients who receive ≥1 dose of study drug and provide ≥1 postdose plasma PK sample.
Morariu-Zamfir:Incyte Corporation: Employment. Bleam:Incyte Corporation: Employment. Yan:Incyte Corporation: Employment.

