

Barth syndrome is an inherited X-linked disorder characterized by cardiomyopathy, skeletal muscle myopathy, and neutropenia. The syndrome arises because of inherited mutations in the gene TAZ, resulting in a loss of function of the protein tafazzin. Of note, a group of investigators recently described how tafazzin can regulate 'stemness' in models of acute myeloid leukemia (Cell Stem Cell, 2019).
Tafazzin is an enzyme that processes the final step of cardiolipin maturation, replacing saturated with unsaturated acyl chains. Cardiolipin is a 4-tailed phospholipid that is almost-exclusively found in the inner membrane of the mitochondria. The lack of tafazzin activity results in a cardiolipin pool that contains more highly saturated lipid tails and it is this lack of unsaturated cardiolipins that contributes to a disorganized inner mitochondrial membrane.
The link between tafazzin-deficiency and myopathy is generally explained by the dependence of muscle cells on mitochondrial function as well as oxidative respiration. The components of the electron transport chain are co-localized with cardiolipin in the inner mitochondrial membrane, and it is felt that their appropriate organization within the membrane lipid bilayer is dependent on the presence of mature cardiolipin which is lacking in those individuals with Barth syndrome.
The link between tafazzin-deficiency and neutropenia is less clear. Neutrophils are terminally-differentiated effector cells of the innate immune system. They are critical for protection against bacterial and fungal pathogens and patients without sufficient neutrophils are among the most immunocompromised and at risk of lethal infection. Neutrophils have few mitochondria at baseline and are generally believed to rely primarily on glycolysis for energy production. It is not known if the mechanism of neutropenia in Barth syndrome is due to a lack of production or due to increased clearance (e.g. more prone to apoptosis).
We undertook the study of tafazzin-deficient neutrophils to try to elucidate the mechanism of neutropenia in patients with Barth syndrome. We took advantage of an existing tafazzin-knockout mouse and a system of conditional immortalization of granulocyte-monocyte progenitors (GMP) using the ER-Hoxb8 system pioneered in our laboratory. This ER-Hoxb8 system allows for the unlimited ex vivo expansion of myeloid progenitors in the presence of estradiol and active Hoxb8. Once estradiol is removed from culture media, the Hoxb8 protein is inactive and the cells undergo normal, synchronous and terminal neutrophilic differentiation. In this manner, we were able to generate tafazzin-wild-type and knockout GMP lines from murine fetal liver cells.
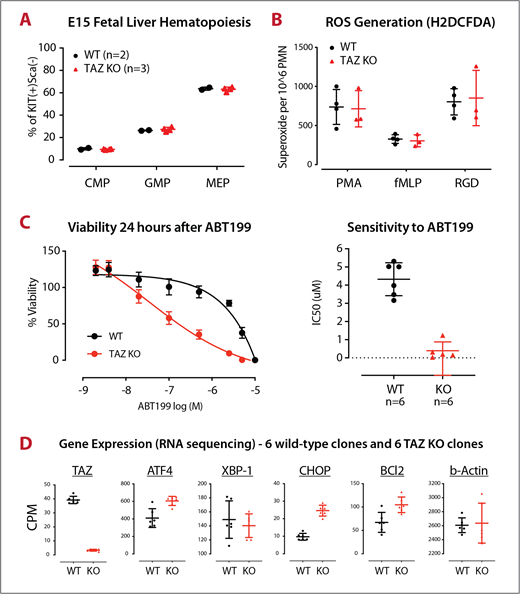
Analysis of the myeloid progenitor compartment in fetal liver cells (d14.5-d16.5) showed no difference between wild-type and knockout mice, arguing against a developmental defect (E15 results shown in PANEL A). Furthermore, the tafazzin-deficient ER-Hoxb8 GMPs and neutrophils were remarkably normal when tested across a variety of assays including phagocytosis, cytokine production and ROS generation (ROS by H2DCFDA shown in PANEL B).
We hypothesized that the unpredictable neutropenia in patients with Barth Syndrome might be due to an increased proclivity to apoptosis because of the mitochondrial membrane defect. Indeed, the tafazzin-deficient GMPs showed an increased sensitivity to Bcl2-inhibition following treatment with ABT199 (PANEL C).
Two lines of evidence have suggested that the increased tendency towards apoptosis may be due to endoplasmic-reticulum (ER) stress. (1) Transmission electron microscopy demonstrated 'swollen' ER in the tafazzin-deficient cells (not shown) and (2) a comparison of gene expression patterns demonstrated an increased expression of ATF4 and CHOP (DDIT3) in the tafazzin-deficient cells (PANEL D).
We are now focused on validating these findings and in establishing models to confirm the ER-stress phenotype in vivo in the TAZ-knockout mouse model as well as primary samples from patients with Barth Syndrome.
We hope that this line of work will confirm the mechanism of neutropenia and shed light on potential targets for therapeutic intervention. In addition, this very rare disorder has provided insight into a previously-unexpected link between neutrophil survival and the membrane integrity of the inner mitochondrial membrane.
Sykes:Clear Creek Bio: Equity Ownership, Other: Co-Founder.

