

Introduction:
Amyloidosis is a disease characterized by deposition of congophilic amyloid fibrils in the extracellular matrix of tissues and organs. Although pulmonary involvement in amyloidosis is common, it is rarely symptomatic. The clinical course is considered indolent or benign, however literature available on biology and history of this disease is limited. Sub-typing of pulmonary amyloidosis has been historically difficult. Pulmonary amyloidosis can be a part of an underlying plasma cell dyscrasia or related to an underlying inflammatory systemic disease. Our aim was to identify the further workup done after a biopsy proven diagnosis of pulmonary amyloidosis and the relation to monoclonal gammopathy. We also investigated the frequency of utilization of mass spectrometry for amyloid typing at our institution.
Methods:
We reviewed medical records of adult patients ≥ 18 years with localized pulmonary amyloidosis from January 1st 2010 to June 30th 2019. The word "amyloidosis" was used to search for cases of localized pulmonary amyloidosis in the hospital's electronic medical records to generate a patient list. We recorded age and gender, medical history, work up for paraproteinemia, renal and hepatic function, pathology results including bone marrow biopsy, imaging studies, treatment and survival.
Results:
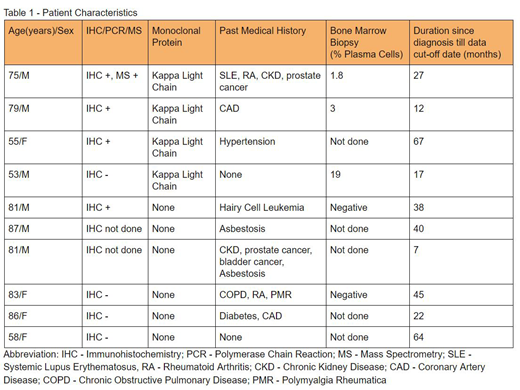
We identified 10 patients with amyloidosis on lung biopsy, out of which 4 were females (40%) and 6 were males (60%). Patient age ranged from 53 to 87 years with a median age of 80 years. (Table - 1)
Four out of the 10 patients were found to have kappa light chains. Out of these 4 patients, 3 had kappa light chain restriction on the amyloid deposit of lung biopsy specimen by immunohistochemistry (IHC). One patient had expression of both kappa and lambda light chains. His B-cell polymerase chain reaction (PCR) was negative. Mass spectrometry was done in 1 patient which confirmed Immunoglobulin light chain (AL) amyloidosis. Bone marrow biopsy was performed in 3 out of 4 patients and interestingly, the patient with negative IHC in the amyloid had 19% plasma cells while the other 2 patients had 1.8% and 3%. All the 4 patients had normal renal function, hepatic function, serum albumin. They also had no bone lesions or organomegaly.
Out of the 10 patients, 1 patient had a history of hairy cell leukemia and the amyloid nodule showed lambda light chain restriction, likely related to the B-cell Lymphoproliferative disorder. He had no evidence of a monoclonal gammopathy or evidence of organ involvement. The remaining 5 patients were suspected to have amyloid A (AA) amyloidosis and 4 of them had documented underlying systemic disease. None of these patients had evidence of other organ involvement.
One patient with suspected AA amyloid nodule had excision of the lesion. All the patients had stable lung nodules and none of them had evidence of progressive systemic amyloidosis. None of the patients were given systemic therapy. Eight out of 10 patients were alive at the time of data cut-off. The 2 deaths were unrelated to amyloidosis were due to hairy cell leukemia in one patient and sepsis secondary to urinary tract infection in the other.
Conclusion:
Our institutional review of localized pulmonary amyloidosis showed no evidence of disease progression and had good clinical outcome. Interestingly, one of our patients with smoldering myeloma with pulmonary amyloid nodule had negative IHC stain which may point towards the fact that these patients may have amyloid which is unrelated to the paraprotein. Even though mass spectrometry is the gold standard for confirmation of amyloid, it is not widely available and may not be indicated in every case. In our study, we found only 10% rate of utilization of mass spectrometry. The other interesting finding was that patients with B-cell lymphoproliferative disorders can have amyloid with light chain restriction which is a part of the disorder rather than AL amyloidosis.
No relevant conflicts of interest to declare.

