

Introduction: Chimeric Antigen Receptor (CAR) based cellular-immunotherapies have demonstrated significant clinical efficacy in haematological malignancies. However, the progress of cellular-immunotherapy for the treatment of Acute Myeloid Leukaemia (AML) has failed to gain momentum due to the lack of targetable tumour specific antigens. CD38 is a transmembrane glycoprotein expressed in lymphoid and myeloid cells with high expression in plasma B-cells, and is a well validated target for anti-CD38 therapy in Myeloma. A recent study has furthermore shown that a proportion of AML patients express CD38 on their leukemic blasts. TNF-related apoptosis-inducing ligand (TRAIL) receptor DR4 is another targetable antigen which has been shown to be expressed in 70% of AML patients. In this study, we investigate the therapeutic efficacy of "affinity-optimized" variant(s) of CD38 CAR and membrane bound TRAIL on NK-cell based platforms which can target AML blasts with high expression of CD38 (CD38high AML). The CAR variant is a CAR which binds with lower affinity to CD38 expressed on healthy immune cells such as CD38positive NK cells, while targeting CD38high AML. The membrane bound TRAIL variant (TRAIL4c9) is a mutant which binds with higher affinity to TRAIL-DR4 on AML cells, whilst avoiding binding to decoy receptors. We hypothesize that genetically modifying NK cells to express "affinity optimized" CD38 CARand/or TRAIL4c9 can effectively eliminate CD38high AML cells.
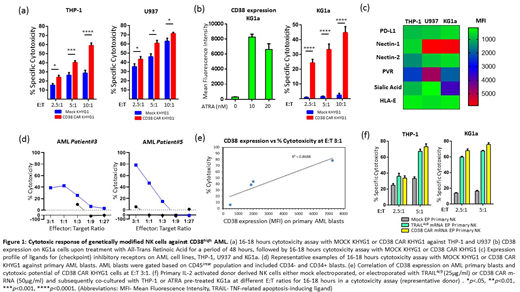
Methods: AML cell lines THP-1, U937, and KG1a were immunophenotyped for CD38 and TRAIL-DR4 expression. Retrovirally transduced CD38 CAR-KHYG1 NK cells were used as immune effector cells and were co-cultured with AML cell lines in cytotoxicity assays. CD38low AML cell line KG1a was pre-treated with 10nM all-trans-retinoic acid (ATRA) to upregulate CD38 expression and were subsequently co-cultured with CD38 CAR-KHYG1 in cytotoxicity assays. CD38 CAR-KHYG1 was also co-cultured with n=4 patient derived AML cells in cytotoxicity assays. Using Maxcyte GT electroporation system primary donor derived IL-2 activated NK cells were either mock electroporated, or electroporated with TRAIL4c9 m-RNA orCD38 CAR m-RNA and subsequently co-cultured with THP-1 or ATRA pre-treated KG1a in a cytotoxicity assay. Expression of pro-apoptotic, anti-apoptotic and ligands for checkpoint inhibitory receptors was analysed by immunoblotting or flowcytometry.
Results: Based on immunophenotyping, we classified AML cell lines as CD38high (THP-1), CD38moderate (U937) and CD38low (KG1a). CD38 CAR-KHYG1 was significantly more cytotoxic than MOCK KHYG1 against CD38high THP-1, at E:T ratios of 2.5:1, 5:1 and 10:1. CD38 CAR-KHYG1 were also more cytotoxic than MOCK KHYG1 against CD38moderate U937 at multiple E:T ratios; albeit the increase in cytotoxicity was at a much lower level in comparison to THP-1 (Fig 1a). Pre-treatment of CD38low KG1a cells with 10nM ATRA upregulated the cell surface expression of CD38, which were subsequently eliminated by CD38 CAR KHYG1 at E:T ratios of 2.5:1, 5:1 and 10:1. KG1a was intrinsically resistant to NK cells as compared to THP-1 and U937 (Fig 1b). This could partly be explained by the high intracellular expression of Bcl-xL, and higher cell surface expression of Nectin-1 and Sialic acid which are the ligands for checkpoint inhibitory receptors CD96 and Siglec-7/9 respectively on NK cell (Fig 1c). CD38 CAR-KHYG1 mounted a potent cytotoxic response against primary CD45intermediate AML blasts (n=4 patients) at multiple E:T ratios, and the extent of CAR induced cytotoxicity correlated with the cell surface CD38 expression on the primary AML blasts (R2=0.87) (Fig 1d,e). TRAIL4c9 or CD38 CAR m-RNA electroporated primary donor-derived NK cells were also potent in eliminating THP-1 and ATRA pre-treated KG1a at multiple E:T ratios (Fig 1f). This demonstrates the potential of therapeutically treating AML patients, with high CD38 expression, with a combination of NK cells expressing "affinity-optimized" CD38 CAR and membrane bound TRAIL variant.
Conclusion: The study demonstrates the therapeutic potential of an "affinity-optimized" CD38 CAR NK cell-based therapy, which can potentially be combined with membrane bound TRAIL expressing NK cells to target CD38high AML. In patients with CD38low expressing AML blasts, patients could be pre-treated with ATRA followed by the combination therapy of CD38 CAR and TRAIL expressing NK cells.
Stikvoort:Onkimmune Ltd., Ireland: Research Funding. Kirkham-McCarthy:Onkimmune Ltd., Ireland: Research Funding. Van De Donk:Janssen Pharmaceuticals: Membership on an entity's Board of Directors or advisory committees, Research Funding; Roche: Membership on an entity's Board of Directors or advisory committees; AMGEN: Membership on an entity's Board of Directors or advisory committees, Research Funding; Celgene Corporation: Membership on an entity's Board of Directors or advisory committees, Research Funding; Bristol-Myers Squibb: Membership on an entity's Board of Directors or advisory committees, Research Funding; Bayer: Membership on an entity's Board of Directors or advisory committees; Servier: Membership on an entity's Board of Directors or advisory committees; Takeda: Membership on an entity's Board of Directors or advisory committees. Mutis:Celgene: Research Funding; Janssen Pharmaceuticals: Research Funding; Amgen: Research Funding; BMS: Research Funding; Novartis: Research Funding; Aduro: Research Funding; Onkimmune: Research Funding. Sarkar:Onkimmune: Research Funding. O'Dwyer:Onkimmune: Equity Ownership, Membership on an entity's Board of Directors or advisory committees, Research Funding; Janssen: Membership on an entity's Board of Directors or advisory committees, Research Funding; GlycoMimetics Inc: Research Funding; AbbVie: Consultancy; BMS: Research Funding.

