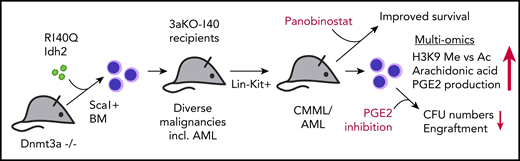
In this issue of Blood, report on their investigation to elucidate the mechanisms by which loss of function of the DNA methyltransferase 3A (DNMT3A) cooperatively induces hematologic malignancies when cooccurring with IDH1/2 mutations.1
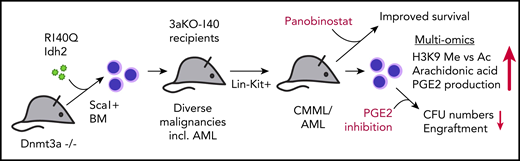
Sca1+ bone marrow (BM) cells from 3aKO mice were retrovirally transduced with R140Q mutated Idh2 (140) and subsequently transplanted in recipient mice in comparison with their single- and nonmutated WT counterpart cells. Recipients of double-mutated (3aKO-140) cells develop severe hematologic malignancies, including AML, earlier than mice receiving 3aKO cells alone. Lin−Kit+ cells harvested from 3aKO-140 injected mice induce CMML or AML in all secondary recipients. Multi-omics approaches reveal an imbalance of H3K9 methylation vs acetylation and overproduction of AA and PGE2. Treatment of mice with the HDACi restores normal levels of H3K9 methylation and acetylation and improves survival of 3aKO-140 injected mice. Ex vivo treatment with celecoxib of 3aKO-140, but not single- or nonmutated progenitors, reduces colony-forming units (CFU) and leukemic engraftment upon retransplantation. incl., including.
Sca1+ bone marrow (BM) cells from 3aKO mice were retrovirally transduced with R140Q mutated Idh2 (140) and subsequently transplanted in recipient mice in comparison with their single- and nonmutated WT counterpart cells. Recipients of double-mutated (3aKO-140) cells develop severe hematologic malignancies, including AML, earlier than mice receiving 3aKO cells alone. Lin−Kit+ cells harvested from 3aKO-140 injected mice induce CMML or AML in all secondary recipients. Multi-omics approaches reveal an imbalance of H3K9 methylation vs acetylation and overproduction of AA and PGE2. Treatment of mice with the HDACi restores normal levels of H3K9 methylation and acetylation and improves survival of 3aKO-140 injected mice. Ex vivo treatment with celecoxib of 3aKO-140, but not single- or nonmutated progenitors, reduces colony-forming units (CFU) and leukemic engraftment upon retransplantation. incl., including.
Acute myeloid leukemia (AML) is a complex disease that regularly involves mutations in several oncogenes and tumor suppressors. Many mutations have been investigated on the single-gene level. However, cooccurring mutations are likely to alter phenotypes, leukemogenic mechanisms, and potentially even drug responses. To model the scenario of combined mutations in DNMT3A and either IDH1/2 as observed in 5% to 8% of patients with myelodysplastic syndrome/AML,2,3 Zhang et al overexpressed the neomorphic Idh2 R140Q mutated protein in hematopoietic progenitors derived from Dnmt3a knockout mice (hereafter called 3aKO-140 cells) and observed development of different hematologic disorders, including overt AML in recipient mice. Using multi-omics approaches, they identified imbalanced H3K9 methylation vs acetylation levels and overproduction of prostaglandin E2 (PGE2) as hallmarks of 3aKO-140 cells. In line with these findings, they found that double-mutant cells from diseased mice were specifically sensitive to histone deacetylase (HDAC) and PGE2 inhibition. The data presented by Zhang et al underline the importance of understanding synergistic mutational events in the development of leukemia. They suggest that the combination of HDAC and prostaglandin synthesis inhibition might represent a novel therapeutic approach for patients with AML harboring both mutations, in particular, for those who develop resistance against available IDH2 inhibitors (see figure).
Patients with AML are currently risk-stratified mostly based on single mutations4 (except for NPM1 and FLT3-ITD). Small molecules targeting mutated proteins, such as FLT35,6 or IDH1/2 inhibitors (reviewed in Golub et al7 ), have shown promising efficacy in patients with AML. So far, most clinical trials assessing the efficacy of compounds targeting mutated proteins usually require only the presence of the respective mutation regardless of comutations. Consequently, the impact of comutations on therapy response is analyzed only retrospectively. Moreover, a substantial fraction of patients will develop resistance against these drugs via diverse mechanisms, including additional mutations in the targeted protein, and will then require alternative therapeutic approaches.
One example of concurrent mutations frequently found in hematologic malignancies involves the genes DNMT3A and IDH1 or IDH2.2,3 Interestingly, DNMT3A and IDH1/2 are antagonizing epigenetic modifiers, as DNMT3A methylates DNA, while IDH1/2 enzymes cause DNA and histone hypomethylation through generation of α-ketoglutarate, an important cofactor of histone and DNA demethylases, such as TET proteins (summarized in Im et al8 ). Consequently, cooccurrence of mutations in DNMT3A and IDH1/2 partially reverses the effects observed when only one of them is mutated.9 This raises the question as to why mutations in both modifiers cooccur in hematologic malignancies and how they cooperate to induce the disease. To address these questions, Zhang et al overexpressed R140Q-mutant Idh2 in hematopoietic progenitor cells harvested from Dnmt3a homozygous KO (3aKO) mice and their wild-type (WT) counterparts. Although a complete loss of the DNMT3A protein is not identical with a heterozygous DNMT3A mutation as observed in the majority of patients, the authors observed that their 3aKO mouse model recapitulates the phenotype of heterozygously mutated Dnmt3a models, such as expansion of hematopoietic stem and progenitor cells (HSPCs).10 Primary recipients of 3aKO-140 cells developed severe hematologic disorders, including overt AML in some recipients with a shorter median survival compared with 3aKO-Idh2 WT mice. Recipients of only Idh2 R140Q engineered HSPCs did not die of hematologic diseases during the observation period. These results clearly demonstrated synergy between the 2 events in inducing hematologic malignancy. To corroborate their findings, they retransplanted the cells in secondary recipients and observed chronic myelomonocytic leukemia (CMML) or AML in all mice injected with Lin-Kit+ cells from primary 3aKO-140 mice.
To gain insight into the mechanism, they performed reduced representation bisulfite sequencing and H3K4me3 and H3K27ac ChIP-seq. These experiments revealed not only increased H3K9 methylation in 3aKO-140 cells but also reduced H3K9 acetylation, which they suggested to be the consequence of competition between enzymes involved in H3K9 methyl- and acetylation. In line with their hypothesis, histone deacetylase inhibitors (HDACi) reversed the observed imbalance and improved survival of mice injected with 3aKO-140 cells. To confirm that they had identified a specific vulnerability of 3aKO-140 cells, they treated single- and double-mutant as well as WT cells with HDACi and inhibitors of H3K9 methylation (G9a inhibitor) and observed specifically high sensitivity of 3aKO-140 cells against these molecules.
To obtain more hints about the cooperative mechanisms, the authors performed a metabolomics approach, which revealed higher abundance of arachidonic acid (AA) in 3aKO-140 cells. Because AA is the precursor of prostaglandin synthesis, they also studied more components of the pathway and found higher levels of PGE2 and overexpression of FADS2 messenger RNA, a key enzyme for AA synthesis, in 3aKO-140 cells, which they suggest to be due in part to loss of enhancer DNA methylation through the loss of Dnmt3a. Given these results, the authors suggested that PGE2 synthesis inhibition should represent another vulnerability of double-mutant cells. To test this, they again treated WT, single, and double 3aKO-140 cells with the PG synthesis inhibitor celecoxib and PTGER antagonists and found specific sensitivity of double-mutant cells toward PGE2 synthesis inhibition.
Together, the paper highlights the need for understanding cooperative events in leukemia development to identify more efficient synergistic treatment options. Their results suggest that patients with AML harboring mutations in both DNMT3A and IDH2 might benefit from combination treatment with HDAC and prostaglandin synthesis inhibitors when IDH2 inhibitors are not applicable. The findings are highly relevant, because HDAC inhibitors as well as prostaglandin synthesis inhibitors are already available in the clinic.
Conflict-of-interest disclosure: C.P. declares no competing financial interests.