

In this issue of Blood, demonstrate that alteration of calcium trafficking in neutrophilic granulocytes due to lack of superoxide generation results in excessive production of leukotriene B4 (LTB4), and they show its key role in the pathogenesis of lung inflammation in chronic granulomatous disease (CGD).1
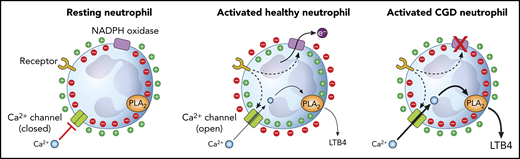
Ion movements in neutrophils initiated by the electrogenic functioning of the NADPH oxidase and alterations in CGD cells. PLA2, phospholipase A2.
Ion movements in neutrophils initiated by the electrogenic functioning of the NADPH oxidase and alterations in CGD cells. PLA2, phospholipase A2.
CGD is an inherited disease caused by a mutation in any of the 5 genes coding for the subunits of the phagocytic nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox2).2 This enzyme transfers 1 electron from cytosolic NADPH to intraphagosomal or extracellular oxygen, which produces superoxide, the precursor for more toxic reactive oxygen species (ROS). ROS deficiency impairs the elimination of several microorganisms leading to severe, recurring, often life-threatening infections.2 However, not all the clinical symptoms can be explained by the lack of superoxide and other ROS.3
About 2 decades ago, we showed that Nox2 deficiency causes altered calcium trafficking in neutrophilic granulocytes (see figure).4 Resting neutrophils have a membrane potential of approximately −60 mV to −80 mV.5 Stimulation of various receptors can initiate electron flow via the assembled Nox2, which removes negative charges from the cytosol.6 In spite of the opening of charge-compensating pathways, mainly H+ channels,5 significant depolarization occurs that can reach positive values up to +58 mV.6,7 This electrophysiological change strongly decreases the driving force for calcium ions and opposes calcium entry via the opening channels. However, in CGD in the absence of a functioning Nox2, activation of neutrophils does not induce dramatic alteration of the driving force, and calcium entry largely exceeds the amount observed in healthy cells.4 After describing the mechanism of enhanced calcium signaling in CGD neutrophils, we speculated on the potential pathogenetic role of the altered ion movements.3,8
The new findings of Song et al provide convincing experimental data on isolated cells and also in a relevant animal model, which confers functional significance to the old observations. Song et al show that CGD neutrophils produce many times more LTB4 (synthesized via the calcium-sensitive cytosolic phospholipase A2α and 5-lipoxygenase [5-LOX]) than healthy cells, and the increased generation of the chemoattractant is proportional to calcium influx. The same results could be achieved by blocking Nox2 in healthy cells and by stimulation of the cells via different receptors. However, the detectable amount of LTB4 was not directly affected by any ROS. In addition, an interesting feed-forward regulation was revealed: neutrophils that have receptors for LTB4 are regulated in an autocrine-paracrine way by this chemoattractant, which augments the difference between CGD and healthy cells. Under in vitro conditions, CGD cells formed large aggregates upon stimulation with zymosan (a yeast membrane extract), and the clusters could be abolished by both an inhibitor of LTB4 synthesis and an antagonist of the LTB4 receptor.
Next, Song et al turned to an animal model and investigated the generation of aberrant lung inflammation induced by the sterile compound zymosan in CGD mice. When zymosan was instilled in the lungs, an increase of neutrophils was detected in the bronchoalveolar lavage fluid and in the lung tissue as early as 8 hours after instillation, and the increase continued up to 24 hours. Accumulation of neutrophils was many times higher in animals with CGD than in wild-type animals. In a similar manner, the concentration of LTB4 increased significantly in CGD animals. By using neutrophil-depleted mice, they verified that neutrophils are responsible for the majority of LTB4 production in the lungs, although other leukocytes are also capable of synthesizing LTB4. To prove the concept, Song et al show that the zymosan-induced hyperinflammation in CGD mice can be almost completely prevented by inhibiting LTB4 synthesis or by blocking LTB4 receptors. With these findings, the important role of neutrophil-derived LTB4 in induction of sterile lung inflammation in CGD mice is clearly supported.
Interestingly, neutrophil recruitment to the lungs was also significantly reduced at 24 hours if the inhibitor of LTB4 synthesis was administered 8 hours after the zymosan treatment. However, if the inhibitor was administered 24 hours after the animal had been exposed to zymosan, it no longer had a protective effect. These findings reveal that LTB4 has a crucial role in development of the aberrant inflammation only in the early period, and they provide another example of the sequential participation of different mediators in neutrophil recruitment.9
The new data communicated by the authors clearly indicate the pathogenetic role of excess LTB4 produced as a result of the enhanced calcium signaling in CGD cells, a process independent of, but adding to, the problem of deficient ROS-related elimination of infectious microbes. However, LTB4 synthesis may not be the only calcium-dependent process that is enhanced because of the missing electrogenic function of Nox2, and may potentially contribute to the complex pathology in CGD.
As with all animal experiments, and specifically in the field of immunology, the relevance to the human disease is a final major question. Aspergillus is a typical microbe that causes serious infections, most often pneumonia with pyogranulomatous infection and abscess formation in patients with CGD2 ; thus, investigation of the effect of yeast cell wall extract is highly relevant. Production of LTB4 is about 10 times higher in murine neutrophils than in human neutrophils, but the supplemental data that accompany the article by Song et al confirm that direction and proportion of changes are the same. LTB4 was found to play an important role in directing recruitment of human neutrophils in general and in pulmonary infiltration in particular. The final proof, however, will come from clinical experience when drugs that attack the LTB4-LTB4 receptor axis or the neutrophil calcium channels are tested in the management of CGD patients.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

