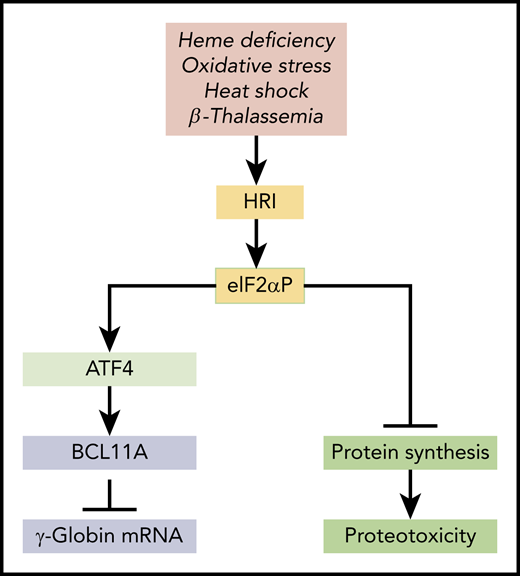
In this issue of Blood, have identified activating transcription factor 4 (ATF4) as a novel regulator of fetal γ-globin gene expression in human cells that promotes BCL11A transcription.1
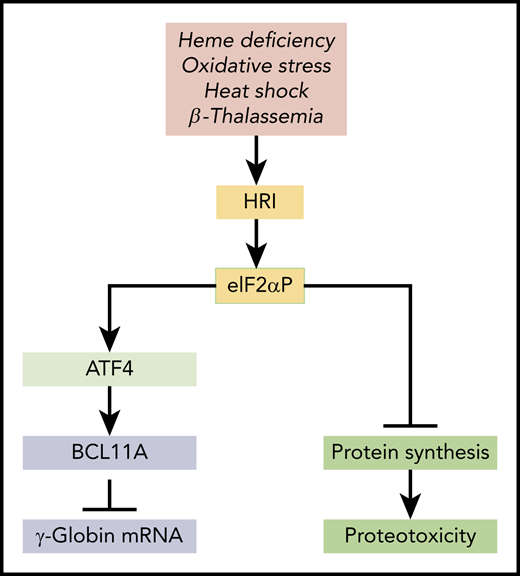
HRI-mediated integrated stress response in HbF expression. HRI is activated in heme deficiency, oxidative stress, heat shock, and pathological conditions of β-thalassemia. Activated HRI phosphorylates its substrate eIF2α, and the phosphorylated eIF2α (eIF2αP) inhibits protein synthesis globally to prevent proteotoxicity incurred during stress. This inhibition of global protein synthesis permits the selective enhancement of the translation of Atf4 mRNA to induce stress response genes for adaptation to stress. The coordinated translational regulation is a universal hallmark across the eIF2α kinase family under various stress conditions and is termed the integrated stress response. Huang et al demonstrated that ATF4 binds to the +55 enhancer of the Bcl11a gene to increase transcription of BCL11A, which represses γ-globin transcription.
HRI-mediated integrated stress response in HbF expression. HRI is activated in heme deficiency, oxidative stress, heat shock, and pathological conditions of β-thalassemia. Activated HRI phosphorylates its substrate eIF2α, and the phosphorylated eIF2α (eIF2αP) inhibits protein synthesis globally to prevent proteotoxicity incurred during stress. This inhibition of global protein synthesis permits the selective enhancement of the translation of Atf4 mRNA to induce stress response genes for adaptation to stress. The coordinated translational regulation is a universal hallmark across the eIF2α kinase family under various stress conditions and is termed the integrated stress response. Huang et al demonstrated that ATF4 binds to the +55 enhancer of the Bcl11a gene to increase transcription of BCL11A, which represses γ-globin transcription.
Earlier work from this group has shown that depletion of heme-regulated inhibitor (HRI), the heme-regulated eIF2α kinase highly expressed in hemoglobinized erythroid cells,2 increases γ-globin gene and thus fetal hemoglobin (HbF) expression in human erythroblasts.3 Persistent HbF expression is known to lessen the severity of β-thalassemia and sickle cell anemia. Approaches in reactivating HbF expression have been pursued widely as potential treatments for β-hemoglobinopathies.4 The article by Huang et al presents a molecular mechanism by which HRI represses γ-globin gene expression by modulating Atf4 messenger RNA (mRNA) translation.
HRI reprograms protein synthesis for adaptive responses to stress. Most notably, HRI is activated in erythroid precursors under iron/heme deficiency, under environmental stresses (eg, oxidative stress, heat shock, and osmatic stress), and under the pathological conditions of β-thalassemia (see figure). Upon activation, HRI phosphorylates the α subunit of eIF2 (eIF2αP), impairing further rounds of ribosome initiation and thereby inhibiting translation. In addition to inhibiting the translation of highly translated mRNAs, eIF2αP also selectively increases translation of certain poorly translated mRNAs for adaptation to stress (see figure).5 This coordinated translational regulation is known as the integrated stress response (ISR). Translational upregulation by eIF2αP requires upstream open reading frames in the 5′ untranslated regions of these unique mRNAs, most notably in the case of Atf4 mRNA. Hri and Atf4 mRNAs are abundantly expressed in erythroblasts.6 In fact, Atf4 mRNA is most highly expressed in hemoglobinized erythroblasts among the 16 differentiation stages of murine bone marrow hematopoietic cells,7 underscoring the significance of HRI-enhanced Atf4 mRNA translation in terminal erythropoiesis.6 Indeed, Atf4−/− mice develop transient fetal anemia8 and exhibit severe anemia in iron deficiency, similarly to Hri−/− mice.9
Interestingly, Huang et al uncovered the role of ATF4 as a negative regulator of HbF expression through an unbiased CRISPR-Cas9 guided loss-of-function screen of transcription factors in HUDEP2 cells, and they confirm this finding in primary cells. They further demonstrated that ATF4 binds to the +55 enhancer of the Bcl11a gene to increase the expression of BCL11A, a repressor of γ-globin gene expression. Importantly, BCL11A expression is reduced upon depletion of ATF4 or removal of the ATF4 binding element within the +55 enhancer. Conversely, overexpression of ATF4 increases BCL11A expression and represses the increased γ-globin gene expression in HRI-depleted cells. This study thus implicates HRI-ISR stress signaling in repressing HbF production through enhanced translational regulation of ATF4 and subsequent upregulation of BCL11A to silence γ-globin gene expression in HUDEP2 cells and primary erythroblasts (see figure). In addition, this study nominates the Bcl11a +55 enhancer as a potential target for therapeutic genome editing.
One surprising aspect in this HRI-ATF4 mediated repression of HbF expression is the species-specific effect observed in human cells but not in mouse cells.1,3 In the Huang et al study, the authors show the conservation of the analogous ATF4 motif in the murine Bcl11a enhancer, and yet it is largely functionally dispensable. HRI deficiency has no effect on BCL11A or γ-globin gene expression in Townes βAβS heterozygote mice, which do not exhibit anemic phenotypes. Because HRI-mediated ATF4 expression is a stress response, it is important to test HRI’s role under stress conditions in homozygote sickle mice. It is possible that the species difference observed is in part the result of the model systems available for investigation in murine and human systems. The in vivo system that uses genetically modified mice is less stressful than the in vitro cell culture conditions used in human studies, which are more stressful and can induce HRI-ATF4 stress response. For example, using the in vitro culture conditions of the murine erythroid progenitors, HRI-ISR is activated even in iron/heme sufficiency,5 but it remains inactive in the bone marrow of mice under iron/heme sufficiency.9 Moreover, murine Hri−/− erythroid progenitors exhibit significant inhibition of erythroid differentiation in vitro,5 whereas Hri−/− mice displayed very mild erythroid phenotypes under iron sufficiency.9
It is important to note that the foremost function of HRI is to inhibit protein synthesis to mitigate proteotoxicity (see figure). It is this inhibitory effect of HRI in protein synthesis that permits the enhanced translation of Atf4 mRNA. Increased ATF4 expression is necessary to mitigate oxidative stress in both human and mouse cells. Hahn and Lowrey10 also reported earlier that HRI-eIF2αP enhanced the translation of γ-globin mRNA. Altogether, HRI kinase seems to have pleiotropic roles in erythropoiesis and hemoglobin synthesis.2
In the future, it will be important to investigate the role of the HRI-ISR signaling in humans under more physiological conditions, for example, by examining the association of Hri or Atf4 genes with HbF production in patients with β-thalassemia or sickle cell anemia. In summary, the Huang et al discovery of ATF4 as a downstream target of HRI stress signaling in silencing γ-globin gene transcription advances our understanding of fetal globin gene expression and connects translational regulation by HRI with downstream functional impacts on transcriptional regulation by ATF4. Furthermore, this study broadens our capabilities to develop new therapeutics for β-hemoglobinopathies.
Conflict-of-interest disclosure: The author declares no competing financial interests.