Key Points
Bhlhe40 is a key transcription factor that regulates CD4+ T cell-mediated GM-CSF production and pathological damage in the colon in GVHD.
CD4+ T-cell–derived GM-CSF production links adaptive to innate immunity by promoting indirect alloantigen presentation in the GI tract.

Abstract
Gastrointestinal (GI) tract involvement is the major cause of morbidity and mortality in acute graft-versus-host disease (GVHD), and pathological damage is largely attributable to inflammatory cytokine production. Recently, granulocyte-macrophage colony stimulating factor (GM-CSF) has been identified as a cytokine that mediates inflammation in the GI tract, but the transcriptional program that governs GM-CSF production and the mechanism by which GM-CSF links adaptive to innate immunity within this tissue site have not been defined. In the current study, we identified Bhlhe40 as a key transcriptional regulator that governs GM-CSF production by CD4+ T cells and mediates pathological damage in the GI tract during GVHD. In addition, we observed that GM-CSF was not regulated by either interleukin 6 (IL-6) or IL-23, which are both potent inducers of GVHD-induced colonic pathology, indicating that GM-CSF constitutes a nonredundant inflammatory pathway in the GI tract. Mechanistically, GM-CSF had no adverse effect on regulatory T-cell reconstitution, but linked adaptive to innate immunity by enhancing the activation of donor-derived dendritic cells in the colon and subsequent accumulation of these cells in the mLNs. In addition, GM-CSF promoted indirect alloantigen presentation, resulting in the accumulation of donor-derived T cells with a proinflammatory cytokine phenotype in the colon. Thus, Bhlhe40+ GM-CSF+ CD4+ T cells constitute a colitogenic T-cell population that promotes indirect alloantigen presentation and pathological damage within the GI tract, positioning GM-CSF as a key regulator of GVHD in the colon and a potential therapeutic target for amelioration of this disease.
Introduction
Graft-versus-host disease (GVHD) is the primary complication after allogeneic hematopoietic stem cell transplantation.1,2 During the acute phase of GVHD, inflammation is restricted to a limited subset of target organs, specifically the skin, liver, and gastrointestinal (GI) tract.3 Compelling data in experimental models have shown that the GI tract plays a pivotal role in the amplification of systemic disease, largely because of the high concentration of proinflammatory pathogen-associated patterns and damage-associated molecular patterns.4 Moreover, damage to the GI tract during GVHD is a primary determinant of clinical outcome in patients with this disease.5,6 Although T cells are the proximate drivers of GVHD,7,8 disease induction and amplification rely on crosstalk between the innate and adaptive arms of the immune system, particularly in the setting of gastrointestinal inflammation.9,10 However, the cellular and cytokine networks that mediate this interplay are incompletely understood.
Granulocyte-macrophage colony stimulating factor (GM-CSF) is a monomeric glycoprotein secreted by a wide array of cell types in both physiological and inflammatory processes.11 Although it plays a limited role in the development of specialized immune subsets,12-14 it is largely redundant with respect to the development of the hematopoietic system.15 Rather, recent work has implicated it as a paracrine-acting proinflammatory cytokine that is able to activate a number of cell populations that comprise the innate immune system (eg, dendritic cells, monocytes, macrophages).16-18 Importantly, this cytokine is produced largely by T cells and then sensed exclusively by myeloid cells,19 positioning GM-CSF as a critical molecule linking the adaptive and innate arms of the immune system. Within the context of GVHD, recent studies have implicated donor-derived GM-CSF as a crucial driver of inflammation and a determinant of disease severity.20,21 However, although GM-CSF is important in the pathophysiology of GVHD, how this cytokine links adaptive to innate immunity and specifically promotes pathological damage in the GI tract has not been clearly defined. The goal of the current study was therefore to define mechanistic pathways by which GM-CSF induces inflammation within this tissue site.
Material and methods
Mice
C57BL/6 (B6) (H-2b), Balb/c (H-2d), GM-CSF−/−, interleukin (IL)-1R−/−, and B6 Foxp3EGFP mice were bred in the Biomedical Resource Center at the Medical College of Wisconsin or purchased from Jackson Laboratories (Bar Harbor, ME). H2-Ab1−/− mice were purchased from Taconic (Rensselaer, NY). Bhlhe40−/− and CD45.1+ TEa mice have been previously described.22,23 Because Bhlhe40−/− mice develop immune dysregulation as they age,24 we employed Bhlhe40−/− donors at 5 to 6 weeks, as there is no evidence of lymphoproliferation, and used wild-type (WT) littermates as controls in transplant experiments. IL-10BiT-Foxp3EGFP reporter mice were provided by Casey Weaver (University of Alabama-Birmingham).25
Other detailed methods
All other methods are described in the supplemental Data, available on the Blood Web site.
Results
The transcription factor Bhlhe40 regulates pathological damage mediated by CD4+ T cells in the colon during GVHD
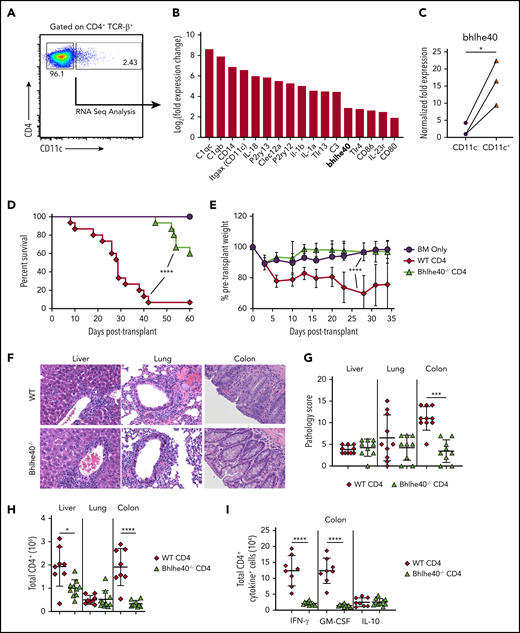
In previous studies, we identified a CD4+ CD11c+ T-cell population that has a biased central memory phenotype, increased expression of the gut-homing molecules α4β7 and CCR9, and an innate-like gene signature, and is important for orchestrating early inflammatory events in the GI tract during GVHD.26 In addition, the pathogenicity of this population was found to be critically dependent on co-expression of the IL-23 receptor. Using an optimized cell sorting approach (supplemental Methods), we compared gene expression profiles of highly purified splenic CD4+ T-cell receptor β+ (TCRβ+) CD11c+ cells with their CD4+ TCRβ+ CD11c− counterparts, using mRNA sequencing to validate our prior findings (Figure 1A). This analysis confirmed that CD4+ TCRβ+ CD11c+ cells, which have the morphological appearance of T lymphocytes (supplemental Figure 1), had increased expression of genes encoding complement (C1qc, C1qb, C3), purinergic receptors (P2ry13, P2ry12), Toll-like receptors (Tlr13, Tlr4), and downstream mediators of damage-associated molecular patterns (IL-1α, IL-1β, IL-18) characteristic of an innate-like signature (Figure 1B; supplemental Table 1). Notably, we observed that there was also increased expression of Bhlhe40 (Figure 1B), which is a transcription factor that has been shown to regulate the production of GM-CSF and promote pathogenicity in murine models of neuroinflammation.22,27 This observation was confirmed by quantitative polymerase chain reaction, which showed increased expression of Bhlhe40 in CD4+ TCRβ+ CD11c+ vs CD4+ TCRβ+ CD11c− T cells (Figure 1C). The identification of Bhlhe40 in this colitogenic T-cell population therefore led us to postulate that this transcription factor might be important in the regulation of colonic inflammation during GVHD.
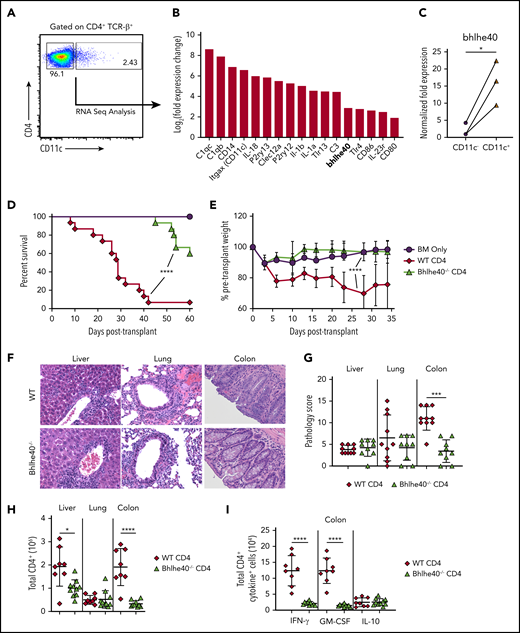
Bhlhe40 regulates CD4+ T-cell-mediated pathological damage in the colon during GVHD. (A) Representative dot plot depicting CD4+ CD11c+ and CD4+ CD11c− T-cell populations that were sorted for subsequent RNA sequence analysis. (B) Waterfall plot showing selected genes from RNA sequencing analysis from sorted splenic CD4+ CD11c+ and CD4+ CD11c− T cells. Data are expressed as log2 (expression in CD4+ TCRβ+ CD11c+/expression in CD4+ TCRβ+ CD11c−) and represent the mean of 3 independent samples. (C) mRNA expression of Bhlhe40 in sorted CD4+ CD11c+ vs CD4+ CD11c− T cells. The expression of 18s rRNA was used as an internal control. Fold change expression was normalized to the median expression of Bhlhe40 over 18s in CD11c− cells. Data are from 3 independent sorted samples of pooled splenocytes from 5 mice. (D, E). Lethally irradiated Balb/c mice were transplanted with Rag-1−/− BM alone (5 × 106) or together with purified CD4+ T cells (1.1 × 106) from bhlhe40−/− or WT sex-matched littermate controls. Overall survival is depicted in panel D, and serial weight curves expressed as a percentage of pretransplant weight in panel E. Results are from 3 independent experiments with 9 to 15 mice per group. (F-I). Irradiated Balb/c mice were transplanted with Rag-1−/− BM alone (5 × 106) plus purified CD4+ T cells (0.9 × 106) from bhlhe40−/− or WT sex-matched littermate controls. (F). Representative hematoxylin and eosin-stained sections of the liver, lung, and colon of transplant recipients from each cohort. Original magnification is 100× for photomicrographs. (G) Pathological scores of liver, lung and colon 21 days posttransplantation, using a semiquantitative scoring system, are depicted. Results are from 2 experiments (n = 9-10/group). (H) Total number of CD4+ T cells isolated from liver, lung, and colon of recipient mice 21 days posttransplantation. (I) Absolute number of CD4+ T cells producing IFN-γ, GM-CSF, and IL-10 in the colon. Results are from 2 experiments for panels H-I (n = 8-9/group). *P < .05; ***P < .001; ****P < .0001.
Bhlhe40 regulates CD4+ T-cell-mediated pathological damage in the colon during GVHD. (A) Representative dot plot depicting CD4+ CD11c+ and CD4+ CD11c− T-cell populations that were sorted for subsequent RNA sequence analysis. (B) Waterfall plot showing selected genes from RNA sequencing analysis from sorted splenic CD4+ CD11c+ and CD4+ CD11c− T cells. Data are expressed as log2 (expression in CD4+ TCRβ+ CD11c+/expression in CD4+ TCRβ+ CD11c−) and represent the mean of 3 independent samples. (C) mRNA expression of Bhlhe40 in sorted CD4+ CD11c+ vs CD4+ CD11c− T cells. The expression of 18s rRNA was used as an internal control. Fold change expression was normalized to the median expression of Bhlhe40 over 18s in CD11c− cells. Data are from 3 independent sorted samples of pooled splenocytes from 5 mice. (D, E). Lethally irradiated Balb/c mice were transplanted with Rag-1−/− BM alone (5 × 106) or together with purified CD4+ T cells (1.1 × 106) from bhlhe40−/− or WT sex-matched littermate controls. Overall survival is depicted in panel D, and serial weight curves expressed as a percentage of pretransplant weight in panel E. Results are from 3 independent experiments with 9 to 15 mice per group. (F-I). Irradiated Balb/c mice were transplanted with Rag-1−/− BM alone (5 × 106) plus purified CD4+ T cells (0.9 × 106) from bhlhe40−/− or WT sex-matched littermate controls. (F). Representative hematoxylin and eosin-stained sections of the liver, lung, and colon of transplant recipients from each cohort. Original magnification is 100× for photomicrographs. (G) Pathological scores of liver, lung and colon 21 days posttransplantation, using a semiquantitative scoring system, are depicted. Results are from 2 experiments (n = 9-10/group). (H) Total number of CD4+ T cells isolated from liver, lung, and colon of recipient mice 21 days posttransplantation. (I) Absolute number of CD4+ T cells producing IFN-γ, GM-CSF, and IL-10 in the colon. Results are from 2 experiments for panels H-I (n = 8-9/group). *P < .05; ***P < .001; ****P < .0001.
In vitro assays revealed that WT and Bhlhe40−/− CD4+ T cells had similar alloantigen-induced proliferation (supplemental Figure 2A-B), but that Bhlhe40−/− CD4+ T cells produced significantly less interferon (IFN)-γ and GM-CSF (supplemental Figure 2C-D). To determine the functional significance of Bhlhe40 expression in CD4+ T cells, we then performed transplantation studies in which recipients were reconstituted with Rag-1−/− bone marrow (BM) alone or together with purified CD4+ T cells from WT or Bhlhe40−/− donors. Animals transplanted with Bhlhe40−/− CD4+ T cells had significantly improved survival and weight recovery when compared with WT control mice (Figure 1D-E), indicating that this transcription factor imparts a proinflammatory phenotype on donor-derived CD4+ T cells. Histological analysis revealed a reduction in overall pathological damage in the colon, but no difference in either the liver or the lung (Figure 1F-G). This was accompanied by a significant decrease in the absolute number of CD4+ T cells in the colon in recipients of Bhlhe40−/− grafts (Figure 1H). Nearly all CD4+ T cells in the colon in both cohorts expressed an effector memory (ie, CD44hi CD62Llo) phenotype (supplemental Figure 3A-B), and there was a very modest increase in the percentage of Bhlhe40−/− CD4+ T cells that expressed Fas ligand (supplemental Figure 3C-D). Intracellular cytokine analysis demonstrated a significant reduction in the absolute number of CD4+ IFN-γ+ and GM-CSF+ T cells in recipients of Bhlhe40−/− grafts, consistent with in vitro results, but no change in CD4+ IL-10+ cells (Figure 1I). Collectively, these studies indicated that Bhlhe40 potentiated CD4+ T-cell-mediated inflammation in the colon during GVHD.
IL-1 is not required forBhlhe40 expression or GM-CSF production in colon-derived CD4+ T cells during GVHD
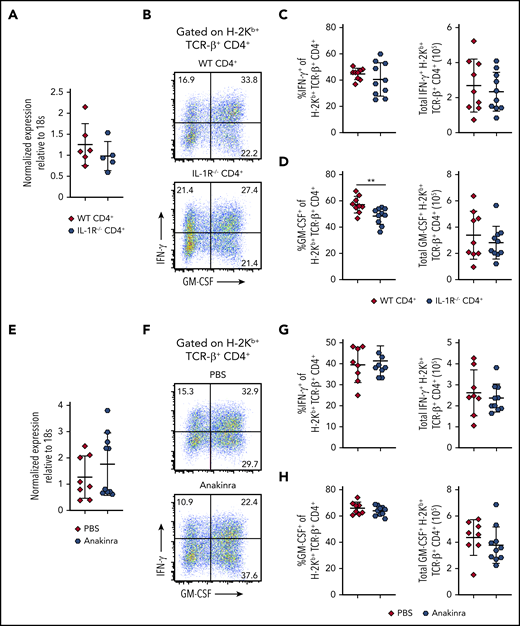
IL-1β signaling through the IL-1R has been shown to induce expression of Bhlhe40 in experimental allergic encephalomyelitis.27 To determine whether IL-1 signaling affected Bhlhe40 expression and GM-CSF production during GVHD, lethally irradiated Balb/c mice were transplanted with Rag-1–/– BM alone or together with CD4+ T cells from either WT or IL-1R−/− animals. We observed that there was no difference in expression of Bhlhe40 in the colons of mice reconstituted with WT vs IL-1R−/− T CD4+ cells (Figure 2A). In addition, absence of the IL-1R on CD4+ T cells resulted in a reduction in the frequency of CD4+ GM-CSF+ T cells, but no difference in the absolute number of CD4+ T cells that produced GM-CSF or IFN-γ (Figure 2B-D). In subsequent experiments, we employed a complementary approach by treating mice with Anakinra, which is a human IL-1R antagonist that was previously shown to have a protective effect in murine GVHD.28 There was no difference in Bhlhe40 expression in CD4+ T cells derived from the colons of mice that were administered Anakinra vs a vehicle control (Figure 2E). Moreover, we observed no difference in the frequency or absolute number of donor-derived CD4+ T cells that produced IFN-γ (Figure 2F-G) or GM-CSF (Figure 2F,H) in this tissue site. Thus, IL-1 did not augment expression of Bhlhe40, nor did it significantly enhance GM-CSF production in colon-derived CD4+ T cells during GVHD.
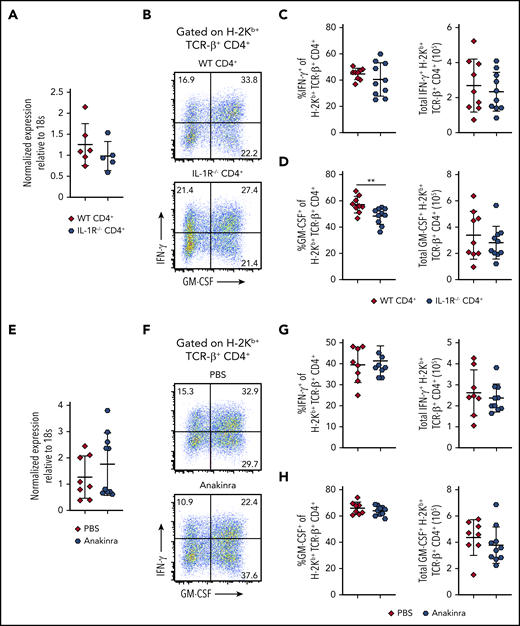
IL-1 is not required for Bhlhe40 expression or GM-CSF production in colon-derived CD4+ T cells during GVHD. (A-D) Lethally irradiated Balb/c mice were transplanted with B6 Rag-1−/− BM (5 × 106) supplemented with 1 × 106 purified CD4+ T cells from WT or IL-1R−/− mice. (A) Bhlhe40 expression in flow-sorted H-2Kb+ TCR-β+ CD4+ cells from the colons of mice 14 days posttransplantation by quantitative reverse transcription polymerase chain reaction is shown. Fold change expression was normalized to the median expression of Bhlhe40 over 18S. Results are from 2 experiments with 5 to 6 mice per group. (B) Representative flow plots gated on H-2Kb+ TCR-β+ CD4+ cells depicting production of GM-CSF and IFN-γ in the colons of mice 14 days posttransplantation. (C-D) The frequency and total number of donor-derived CD4+ T cells in the colon producing IFN-γ (C) or GM-CSF (D). Results are from 2 experiments with 9 to 10 mice/group. (E-H) Lethally irradiated Balb/c mice were transplanted with B6 BM (5 × 106) supplemented with 1 × 106 purified CD4+ T cells from WT mice. Recipient mice received intraperitoneal injections of either Anakinra or PBS, as described in the supplemental Methods. (E) Bhlhe40 expression in flow-sorted H-2Kb+ TCR-β+ CD4+ cells from the colons of mice 14 days posttransplantation by quantitative polymerase chain reaction is shown. Fold change expression was normalized to the median expression of Bhlhe40 over 18S. Results are from 2 experiments with 8 to 10 mice/group. (F) Representative dot plots gated on H-2Kb+ TCR-β+ CD4+ depicting expression of GM-CSF and IFN-γ in the colons of mice 14 days posttransplantation. (G-H) The frequency and total number of donor-derived CD4+ T cells producing IFN-γ (G) or GM-CSF (H). Results are from 2 experiments with 9 to 10 mice/group. **P < .01.
IL-1 is not required for Bhlhe40 expression or GM-CSF production in colon-derived CD4+ T cells during GVHD. (A-D) Lethally irradiated Balb/c mice were transplanted with B6 Rag-1−/− BM (5 × 106) supplemented with 1 × 106 purified CD4+ T cells from WT or IL-1R−/− mice. (A) Bhlhe40 expression in flow-sorted H-2Kb+ TCR-β+ CD4+ cells from the colons of mice 14 days posttransplantation by quantitative reverse transcription polymerase chain reaction is shown. Fold change expression was normalized to the median expression of Bhlhe40 over 18S. Results are from 2 experiments with 5 to 6 mice per group. (B) Representative flow plots gated on H-2Kb+ TCR-β+ CD4+ cells depicting production of GM-CSF and IFN-γ in the colons of mice 14 days posttransplantation. (C-D) The frequency and total number of donor-derived CD4+ T cells in the colon producing IFN-γ (C) or GM-CSF (D). Results are from 2 experiments with 9 to 10 mice/group. (E-H) Lethally irradiated Balb/c mice were transplanted with B6 BM (5 × 106) supplemented with 1 × 106 purified CD4+ T cells from WT mice. Recipient mice received intraperitoneal injections of either Anakinra or PBS, as described in the supplemental Methods. (E) Bhlhe40 expression in flow-sorted H-2Kb+ TCR-β+ CD4+ cells from the colons of mice 14 days posttransplantation by quantitative polymerase chain reaction is shown. Fold change expression was normalized to the median expression of Bhlhe40 over 18S. Results are from 2 experiments with 8 to 10 mice/group. (F) Representative dot plots gated on H-2Kb+ TCR-β+ CD4+ depicting expression of GM-CSF and IFN-γ in the colons of mice 14 days posttransplantation. (G-H) The frequency and total number of donor-derived CD4+ T cells producing IFN-γ (G) or GM-CSF (H). Results are from 2 experiments with 9 to 10 mice/group. **P < .01.
Donor T-cell–derived GM-CSF drives acute GVHD lethality and pathological damage in the GI tract
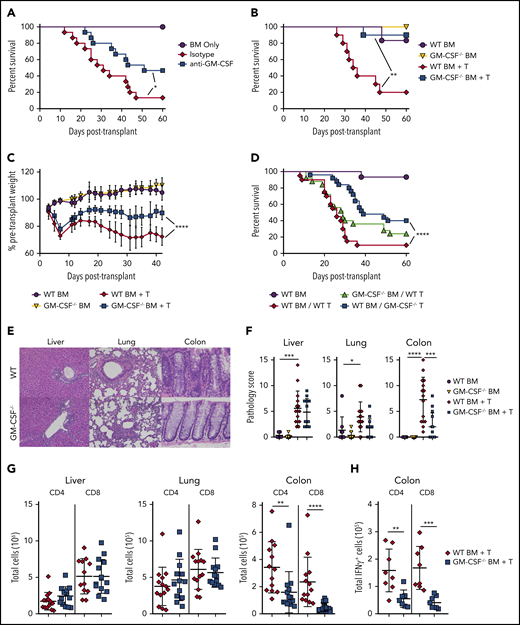
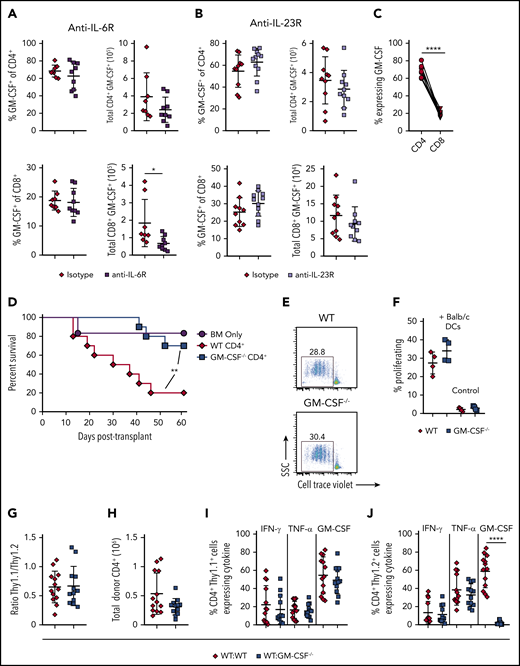
Bhlhe40 is known to positively regulate the production of GM-CSF and reciprocally inhibit IL-10.22 However, given that the total number of CD4+ IL-10+ cells was similar in mice receiving WT or Bhlhe40−/− T cells (Figure 1I), we concluded that the suppression of IL-10 production was not a likely mechanism by which Bhlhe40 induced pathology in gastrointestinal GVHD. This led us to postulate that the proinflammatory effects conferred by expression of Bhlhe40 in CD4+ T cells were attributable, at least in part, to the production of GM-CSF. To formally define the functional role of GM-CSF, we employed antibody and genetic-based approaches to examine the effect of this signaling pathway on GVHD severity. We observed that recipients treated with an anti-GM-CSF antibody had significantly improved overall survival relative to isotype antibody-treated control animals (Figure 3A). Subsequent experiments in which mice were reconstituted with marrow grafts from either WT or GM-CSF−/− animals also revealed that recipients of GM-CSF−/− grafts had significantly increased survival and reduced weight loss when compared with WT controls (Figure 3B-C). Studies to delineate the critical donor GM-CSF-expressing population demonstrated that survival was significantly prolonged in animals transplanted with GM-CSF−/− T cells, whereas mortality was unaffected when GM-CSF expression was absent only from BM-derived cells (Figure 3D), indicating that T-cell expression of GM-CSF was critical. Histological analysis of GVHD target organs demonstrated a significant reduction in pathology scores in the colon of animals that received GM-CSF−/− grafts (Figure 3E-F). There was no difference in the liver or lung, indicative of a preferential protective effect in the GI tract. We also observed a significant reduction in the absolute number of donor-derived CD4+ and CD8+ T cells (Figure 3G), as well as CD4+IFN-γ+ and CD8+IFN-γ+ T cells in the colon (Figure 3H), whereas there was no difference in total CD4+ and CD8+ T cells in the lung or liver. Collectively, these studies demonstrated that T-cell production of GM-CSF exacerbated GVHD by inducing preferential pathological damage in the GI tract.
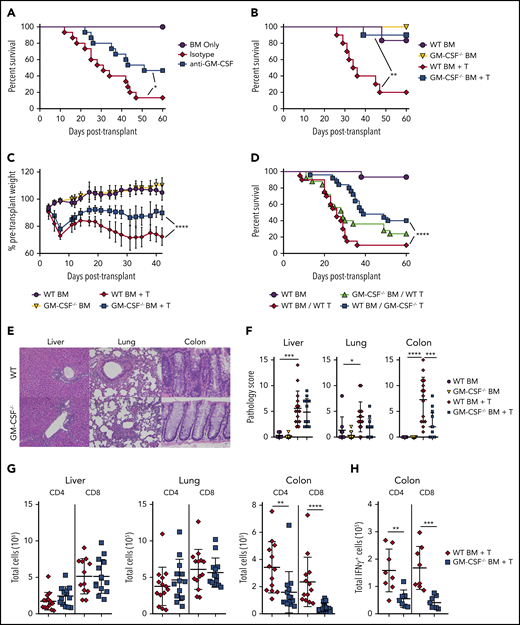
Donor T-cell–derived GM-CSF drives lethal GVHD. (A) Lethally irradiated Balb/c mice were transplanted with B6 BM alone (n = 9) or together with B6 spleen cells (adjusted to yield an αβ+ T-cell dose of 0.85 × 106). Animals receiving adjunctive splenocytes were treated intraperitoneally with 0.5 mg rat IgG (n = 15) or anti-GM-CSF antibody (M250; n = 15) weekly for 5 weeks. Results are from 3 experiments. (B-C) Irradiated Balb/c were transplanted with BM alone from B6 (n = 6) or GM-CSF−/− (n = 6) donors or BM and spleen cells from B6 (n = 10) or GM-CSF−/− animals (n = 10). Overall survival is depicted in panel B, and serial weight curves expressed as a percentage of pretransplant weight in panel C. Results are from 2 experiments. (D) Balb/c mice were transplanted with B6 BM cells (n = 15), B6 BM plus B6 spleen cells (adjusted to yield an αβ+ T cell dose of 0.85 × 106; n = 20), B6 BM plus an equivalent number of GM-CSF−/− spleen cells (n = 25), or GM-CSF−/− BM and B6 spleen cells (n = 25). Overall survival is shown. Results are cumulative results from 5 experiments. (E-H) Balb/c mice were transplanted with BM and spleen cells (adjusted to yield an αβ+ T cell dose of 0.8 × 106) from B6 or GM-CSF−/− mice. Balb/c mice transplanted with B6 BM only served as controls. (E) Representative hematoxylin and eosin-stained sections of the liver, lung, and colon of transplant recipients from each cohort. Original magnification is 100× for photomicrographs. (F) Pathological scores of liver, lung, and colon tissue harvested from animals 21 days posttransplantation using a semiquantitative scoring system are depicted. Data are from 3 experiments with 9 to 15 mice/group. (G) The total number of donor-derived CD4+ or CD8+ T cells isolated from the liver, lung, and colon is shown on day 21 posttransplantation. Data are from 3 independent experiments (n = 13-15/group). (H) The total number of donor-derived CD4+ IFN-γ+ or CD8+ IFN-γ+ T cells in the colon 21 days posttransplantation. Results are from 2 experiments (n = 8 mice/group). *P < .05; **P < .01; ***P < .001; ****P < .0001.
Donor T-cell–derived GM-CSF drives lethal GVHD. (A) Lethally irradiated Balb/c mice were transplanted with B6 BM alone (n = 9) or together with B6 spleen cells (adjusted to yield an αβ+ T-cell dose of 0.85 × 106). Animals receiving adjunctive splenocytes were treated intraperitoneally with 0.5 mg rat IgG (n = 15) or anti-GM-CSF antibody (M250; n = 15) weekly for 5 weeks. Results are from 3 experiments. (B-C) Irradiated Balb/c were transplanted with BM alone from B6 (n = 6) or GM-CSF−/− (n = 6) donors or BM and spleen cells from B6 (n = 10) or GM-CSF−/− animals (n = 10). Overall survival is depicted in panel B, and serial weight curves expressed as a percentage of pretransplant weight in panel C. Results are from 2 experiments. (D) Balb/c mice were transplanted with B6 BM cells (n = 15), B6 BM plus B6 spleen cells (adjusted to yield an αβ+ T cell dose of 0.85 × 106; n = 20), B6 BM plus an equivalent number of GM-CSF−/− spleen cells (n = 25), or GM-CSF−/− BM and B6 spleen cells (n = 25). Overall survival is shown. Results are cumulative results from 5 experiments. (E-H) Balb/c mice were transplanted with BM and spleen cells (adjusted to yield an αβ+ T cell dose of 0.8 × 106) from B6 or GM-CSF−/− mice. Balb/c mice transplanted with B6 BM only served as controls. (E) Representative hematoxylin and eosin-stained sections of the liver, lung, and colon of transplant recipients from each cohort. Original magnification is 100× for photomicrographs. (F) Pathological scores of liver, lung, and colon tissue harvested from animals 21 days posttransplantation using a semiquantitative scoring system are depicted. Data are from 3 experiments with 9 to 15 mice/group. (G) The total number of donor-derived CD4+ or CD8+ T cells isolated from the liver, lung, and colon is shown on day 21 posttransplantation. Data are from 3 independent experiments (n = 13-15/group). (H) The total number of donor-derived CD4+ IFN-γ+ or CD8+ IFN-γ+ T cells in the colon 21 days posttransplantation. Results are from 2 experiments (n = 8 mice/group). *P < .05; **P < .01; ***P < .001; ****P < .0001.
CD4+ T-cell–derived GM-CSF induces pathological damage in the colon and is independent of IL-23 and IL-6 signaling
Prior studies have shown that blockade of either IL-6 or IL-23 signaling is able to specifically reduce inflammation in the colon during GVHD and improve overall survival.29-32 These data prompted us to examine whether either cytokine played an upstream role in regulating the accumulation of GM-CSF-expressing T cells within the GI tract. We observed that there was no difference in the frequency or absolute number of CD4+ or CD8+ T cells that produced GM-CSF in animals treated with either anti-IL-6R (Figure 4A) or anti-IL-23R (Figure 4B) antibodies, except for a small reduction in the total number of CD8+ GM-CSF+ cells in anti-IL-6R-treated animals when compared with mice administered an isotype control antibody. Overall, we observed that there was little CD8+ T-cell production of GM-CSF in isotype antibody-treated animals (Figure 4C), indicating that CD4+ T cells are the primary producers of this cytokine during GVHD. Moreover, CD4+ T-cell–derived GM-CSF production alone was sufficient to drive GVHD lethality in the absence of CD8+ T cells (Figure 4D). In vitro studies revealed that CD4+ T cells from GM-CSF−/− mice proliferated equally well to alloantigen when compared with WT T cells (Figure 4E-F), indicating that these cells were not compromised in their ability to expand. Finally, we examined whether CD4+ GM-CSF−/− T cells were intrinsically defective in their ability to manifest a proinflammatory cytokine phenotype. Using Thy-1 allelic differences to distinguish Thy1.1 B6.PL (GM-CSF+/+) from Thy 1.2 WT (GM-CSF+/+) or GM-CSF−/− CD4+ T cells, we reconstituted mice with either an equal mix of either B6.PL and WT CD4+ or B6.PL and GM-CSF−/− CD4+ T cells. We observed that GM-CSF−/− CD4+ T cells exhibited a similar capacity to expand in the colon of recipient mice (Figure 4G-H) and secrete IFN-γ and tumor necrosis factor (TNF)-α (Figure 4I-J), as WT CD4+ T cells. An increase in the proportion of GM-CSF−/− T cells from 50% to 75% (and a commensurate decrease in WT T cells), however, resulted in a significant decrease in the absolute number of total and Thy1.2+ CD4+ T cells, indicative of a dose effect whereby a threshold number of WT CD4+ T cells was necessary for full inflammatory effects to be observed (supplemental Figure 4). Collectively, these studies indicated that inflammation mediated by GM-CSF was attributable primarily to CD4+ GM-CSF-expressing T cells that were not dependent on either IL-6 or IL-23, and that could drive the differentiation of CD4+ GM-CSF−/− T cells into a proinflammatory phenotype.
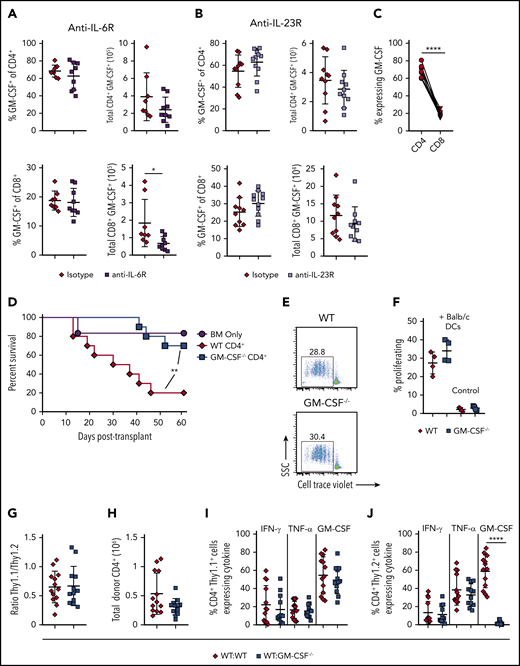
CD4+ T-cell–derived GM-CSF drives inflammation in the colon during GVHD independent of IL-6 and IL-23. (A-B) Lethally irradiated Balb/c mice were transplanted with B6 BM and B6 spleen cells (adjusted to yield an αβ+ T cell dose of 0.8 × 106). Animals were treated intraperitoneally on a weekly basis with an anti-IL-6R (panel A) or anti-IL-23R (panel B) antibody or an appropriate isotype control (n = 8-11/group). The frequency and absolute number of CD4+ GM-CSF+ and CD8+ GM-CSF+ T cells in the colon are shown for each antibody-treated cohort 3 weeks post transplantation. Data are from 2 experiments for anti-IL-6R and 3 experiments for anti-IL-23R. (C) Percentage of GM-CSF expressing donor-derived CD4+ and CD8+ T cells in the colon of mice transplanted as in panel A that were treated with an isotype control antibody. Data were compared using a paired t test. (D) Balb/c mice were transplanted with Rag-1−/− BM alone (5 × 106; n = 6) or together with purified CD4+ T cells (1.1 × 106) from WT (n = 10) or GM-CSF−/− (n = 10) mice. Results are from 2 experiments. (E-F) Magnetically purified CD4+ T cells (1 × 105) from WT or GM-CSF−/− mice were labeled with Cell Trace Violet and cultured with allogeneic Balb/c CD11c-enriched dendritic cells (DCs; 5 × 104) for 3 days. Representative flow plots gated on CD4+ H2-Kb+ cells showing cell trace dye dilution is shown in panel E. The percentage of Cell Tracelo CD4+ T cells in the presence or absence of DCs from replicate experiments (n = 4) is depicted in panel F. (G-J) Balb/c mice were transplanted with B6 Rag-1−/− BM (5 × 106) plus 0.4 × 106 CD4+ T cells in a 1:1 ratio from B6.PL GM-CSF+/+ and B6 GM-CSF−/− animals (WT/ GM-CSF−/−), or B6 Rag-1−/− BM and a mixture of CD4+ T cells from B6.PL GM-CSF+/+ and B6 GM-CSF+/+ mice (WT/WT). Ratio of CD4+ Thy1.1/Thy1.2+ T cells in the colons of mice 21 days posttransplantation is depicted in panel G, and the total number of donor-derived T cells (Thy1.1+ and Thy1.2+) is shown in H. The percentage of CD4+ Thy1.1+ and Thy1.2+ T cells that produced IFN-γ, TNF-α, and GM-CSF is shown in panels I and J, respectively. Data are from 3 experiments with 13 to 15 mice/group. **P < .01; ****P < .0001.
CD4+ T-cell–derived GM-CSF drives inflammation in the colon during GVHD independent of IL-6 and IL-23. (A-B) Lethally irradiated Balb/c mice were transplanted with B6 BM and B6 spleen cells (adjusted to yield an αβ+ T cell dose of 0.8 × 106). Animals were treated intraperitoneally on a weekly basis with an anti-IL-6R (panel A) or anti-IL-23R (panel B) antibody or an appropriate isotype control (n = 8-11/group). The frequency and absolute number of CD4+ GM-CSF+ and CD8+ GM-CSF+ T cells in the colon are shown for each antibody-treated cohort 3 weeks post transplantation. Data are from 2 experiments for anti-IL-6R and 3 experiments for anti-IL-23R. (C) Percentage of GM-CSF expressing donor-derived CD4+ and CD8+ T cells in the colon of mice transplanted as in panel A that were treated with an isotype control antibody. Data were compared using a paired t test. (D) Balb/c mice were transplanted with Rag-1−/− BM alone (5 × 106; n = 6) or together with purified CD4+ T cells (1.1 × 106) from WT (n = 10) or GM-CSF−/− (n = 10) mice. Results are from 2 experiments. (E-F) Magnetically purified CD4+ T cells (1 × 105) from WT or GM-CSF−/− mice were labeled with Cell Trace Violet and cultured with allogeneic Balb/c CD11c-enriched dendritic cells (DCs; 5 × 104) for 3 days. Representative flow plots gated on CD4+ H2-Kb+ cells showing cell trace dye dilution is shown in panel E. The percentage of Cell Tracelo CD4+ T cells in the presence or absence of DCs from replicate experiments (n = 4) is depicted in panel F. (G-J) Balb/c mice were transplanted with B6 Rag-1−/− BM (5 × 106) plus 0.4 × 106 CD4+ T cells in a 1:1 ratio from B6.PL GM-CSF+/+ and B6 GM-CSF−/− animals (WT/ GM-CSF−/−), or B6 Rag-1−/− BM and a mixture of CD4+ T cells from B6.PL GM-CSF+/+ and B6 GM-CSF+/+ mice (WT/WT). Ratio of CD4+ Thy1.1/Thy1.2+ T cells in the colons of mice 21 days posttransplantation is depicted in panel G, and the total number of donor-derived T cells (Thy1.1+ and Thy1.2+) is shown in H. The percentage of CD4+ Thy1.1+ and Thy1.2+ T cells that produced IFN-γ, TNF-α, and GM-CSF is shown in panels I and J, respectively. Data are from 3 experiments with 13 to 15 mice/group. **P < .01; ****P < .0001.
GM-CSF does not affect Treg reconstitution during GVHD
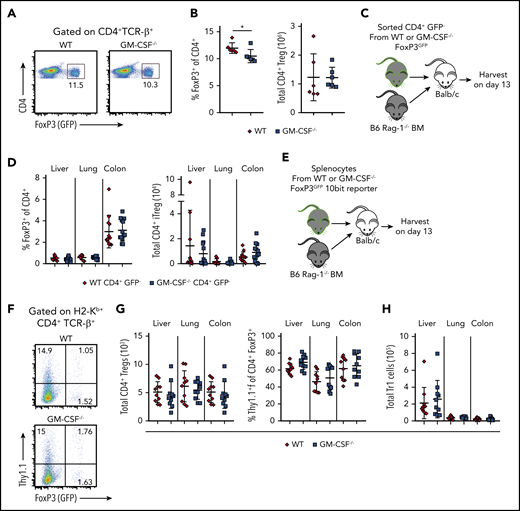
Both CD4+ natural Tregs (nTregs) and induced Tregs (iTregs) play an important role in attenuating GVHD-induced inflammation,33-35 and their reconstitution and stability are impaired in the setting of a proinflammatory environment.35,36 Given the inflammatory role that GM-CSF has in GVHD biology, one possible mechanism by which this cytokine could potentiate T-cell alloreactivity is by inhibiting the reconstitution of Tregs. To address this question, we crossed Foxp3EGFP and GM-CSF−/− mice to create a reporter strain for Tregs that were GM-CSF-deficient. At baseline, there was a slight decrease in the frequency of Tregs in the spleens of Foxp3EGFP GM-CSF−/− mice (mean, 10.5% vs 12%), but no difference in the absolute number (Figure 5A-B). To determine whether GM-CSF affected the induction of Tregs from the conventional T-cell compartment, recipients were transplanted with B6 Rag-1 BM alone or Rag-1 BM plus sorted CD4+ GFP− cells from Foxp3EGFP or Foxp3EGFP GM-CSF−/− mice (Figure 5C). We observed no difference in the frequency or absolute number of iTregs in the liver, lung, or colon of animals reconstituted with either CD4+ T-cell population (Figure 5D). We then assessed whether GM-CSF affected the reconstitution of nTregs or the production of IL-10, which is one of the mechanisms by which these cells suppress GVHD.37 To address this question, we crossed IL-10Bit (Thy1.1) Foxp3EGFP reporter mice with GM-CSF−/− animals and used these mice as donors in transplantation studies (Figure 5E). These studies revealed that the absence of GM-CSF had no effect on the frequency of IL-10-producing Tregs or the absolute number of total nTregs in any of these tissue sites (Figure 5F-G). Prior studies have shown that the preponderance of donor-derived IL-10 production is from CD4+ non-Foxp3-expressing cells (ie, Tr1 cells),26 which can also suppress GVHD.38 However, we observed that there was also no difference in the absolute number of Tr1 cells in any GVHD target tissue (Figure 5H). Collectively, these studies demonstrated that GM-CSF production by CD4+ T cells did not compromise CD4+ Treg reconstitution, indicating that this was not a mechanistic pathway by which GM-CSF exacerbated gastrointestinal tract GVHD.
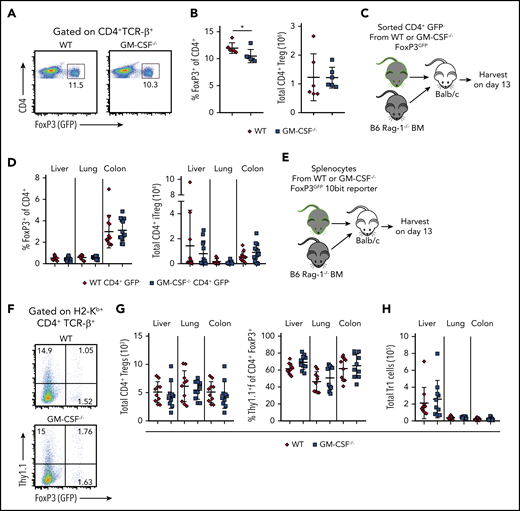
GM-CSF does not adversely affect Treg reconstitution during GVHD. (A) Representative dot plot depicting the percentage of CD4+Foxp3+ (GFP+) Tregs in the spleen of Foxp3EGFP vs GM-CSF−/− Foxp3EGFP mice. (B) Frequency and absolute number of CD4+ Tregs in spleens of Foxp3EGFP vs GM-CSF−/− Foxp3EGFP animals (n = 6 for each genotype). (C-D) TCR-β+ CD4+ GFP− cells (5 × 105) from WT or GM-CSF−/− FoxP3EGFP reporter animals were sort purified and transplanted along with 5 × 106Rag-1−/− BM cells into lethally irradiated Balb/c mice. (C) Experimental design. (D) The frequency and absolute number of H2-Kb+ TCR-β+ CD4+ EGFP+ induced Tregs in the liver, lung, and colon of mice 13 days posttransplantation. Data are from 3 independent experiments (n = 9-14 mice/group). (E-H) Lethally irradiated Balb/c mice were transplanted with 5 × 106 BM cells plus spleen cells (adjusted to yield an αβ+ T cell dose of 0.8 × 106) from WT or GM-CSF−/− 10bit/FoxP3EGFP double reporter mice. (E) Experimental design. (F) Representative dot plot depicting donor-derived CD4+ T cells in the colon that expressed FoxP3 (GFP) and/or IL-10 (Thy1.1). (G) The absolute number of CD4+ FoxP3+ Tregs in the liver, lung, and colon of mice. (H) The absolute number of CD4+ FoxP3− cells which expressed IL-10 (Tr1 cells) in these same tissues. Data in panels G and H are from 2 independent experiments (n = 9-10 per group). *P < .05.
GM-CSF does not adversely affect Treg reconstitution during GVHD. (A) Representative dot plot depicting the percentage of CD4+Foxp3+ (GFP+) Tregs in the spleen of Foxp3EGFP vs GM-CSF−/− Foxp3EGFP mice. (B) Frequency and absolute number of CD4+ Tregs in spleens of Foxp3EGFP vs GM-CSF−/− Foxp3EGFP animals (n = 6 for each genotype). (C-D) TCR-β+ CD4+ GFP− cells (5 × 105) from WT or GM-CSF−/− FoxP3EGFP reporter animals were sort purified and transplanted along with 5 × 106Rag-1−/− BM cells into lethally irradiated Balb/c mice. (C) Experimental design. (D) The frequency and absolute number of H2-Kb+ TCR-β+ CD4+ EGFP+ induced Tregs in the liver, lung, and colon of mice 13 days posttransplantation. Data are from 3 independent experiments (n = 9-14 mice/group). (E-H) Lethally irradiated Balb/c mice were transplanted with 5 × 106 BM cells plus spleen cells (adjusted to yield an αβ+ T cell dose of 0.8 × 106) from WT or GM-CSF−/− 10bit/FoxP3EGFP double reporter mice. (E) Experimental design. (F) Representative dot plot depicting donor-derived CD4+ T cells in the colon that expressed FoxP3 (GFP) and/or IL-10 (Thy1.1). (G) The absolute number of CD4+ FoxP3+ Tregs in the liver, lung, and colon of mice. (H) The absolute number of CD4+ FoxP3− cells which expressed IL-10 (Tr1 cells) in these same tissues. Data in panels G and H are from 2 independent experiments (n = 9-10 per group). *P < .05.
GM-CSF induces the activation and expression of IL-23 by donor-derived, colon-resident, conventional dendritic cells
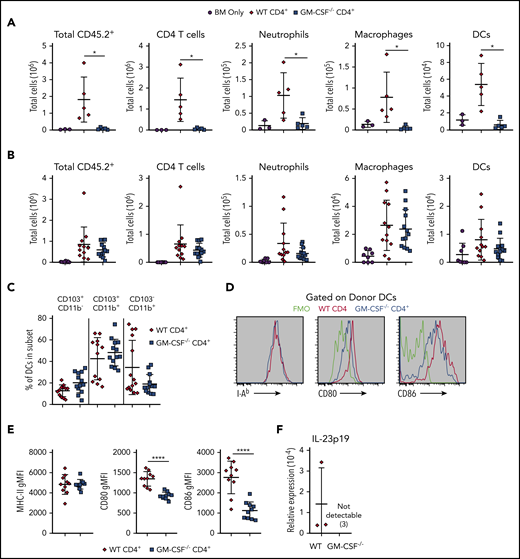
Because T cells do not express the GM-CSF receptor,19 we reasoned that the proinflammatory effects of this cytokine were mediated through GM-CSF-sensitive populations, which we postulated were myeloid cells. To identify GM-CSF–responsive myeloid cells in the colon, animals were transplanted with either Rag-1−/− BM alone or Rag-1−/− BM plus purified CD4+ T cells from WT or GM-CSF−/− mice. The gating strategy to detect specific myeloid cell populations in the colon is shown in supplemental Figure 5. At 3 weeks posttransplantation, which coincided with observed pathological differences in the colon (Figure 3E-F), there was a significant increase in the absolute number of donor-derived neutrophils, macrophages, and dendritic cells (DCs) in the colons of mice transplanted with WT grafts, whereas animals reconstituted with CD4+ GM-CSF−/− T cells had negligible inflammatory cell accumulation (Figure 6A). Thus, T-cell–derived GM‐CSF promoted the accumulation of myeloid cells in the colon. However, the near-complete lack of inflammation in mice transplanted with GM-CSF−/− grafts precluded a detailed analysis of the myeloid compartment in these recipients. Consequently, to uncover a potential mechanism by which GM-CSF-induced myeloid cell expansion and promoted pathological damage, we examined animals at an earlier time (ie, at 2 weeks), as this was the precise point when weight curves of mice receiving WT and GM-CSF grafts diverged (Figure 3C), suggesting that this might represent an inflection point in the disease process when GM-CSF becomes a dominant driver of pathology. In addition, this is also when full donor myeloid reconstitution is typically first established.39 Analysis of mice at this point demonstrated no difference in the absolute number of donor-derived neutrophils, macrophages, or DCs in the colons of mice reconstituted with WT vs GM-CSF−/− CD4+ T cells (Figure 6B). With respect to DCs, the percentages of CD11b− CD103+, CD11b+ CD103+, and CD11b+ CD103− populations were also similar between groups (Figure 6C). However, examination of cell surface markers on DCs revealed that although there was no difference in expression of MHC class II, GM-CSF production by CD4+ T cells resulted in increased expression of the costimulatory molecules CD80 and CD86 (Figure 6D-E). Because IL-23 production by antigen-presenting cells has previously been shown to be partially dependent on GM-CSF,40 we also examined whether DCs in the colon expressed IL-23. Examination of sorted donor-derived conventional DCs (CD45.2+ H2-Kb+ CD64− CD11c+ MHC-II+) revealed that there was detectable expression of Il23a (Figure 6F), but not Il12a (data not shown), in recipients of WT grafts, whereas animals reconstituted with GM-CSF−/− CD4+ T cells had no detectable Il23a or Il12a (data not shown). We observed the macrophages isolated from the colon also had increased expression of CD80 and IL-1β (supplemental Figure 6), indicating that GM-CSF affected other myeloid cells as well. These results demonstrated that CD4+ T-cell–derived GM-CSF augmented myeloid cell accumulation and specifically promoted an activated DC phenotype in the colon characterized by increased expression of costimulatory molecules and IL-23.
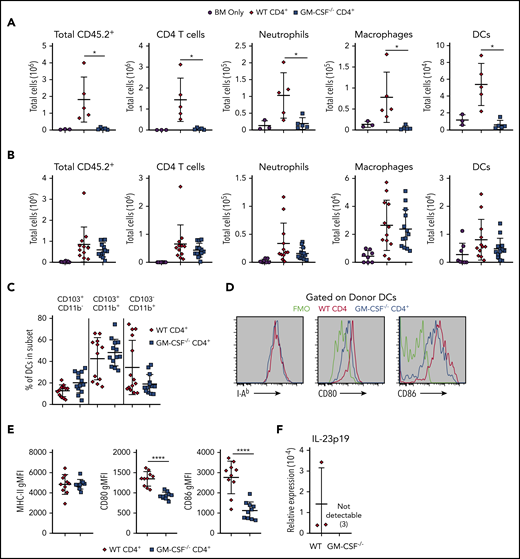
Donor CD4+ T-cell–derived GM-CSF promotes the activation of colonic dendritic cells. (A-E) Lethally irradiated Balb/c mice were transplanted with B6 Rag-1−/− BM (5 × 106) alone or together with 1 × 106 purified CD4+ T cells from WT or GM-CSF−/− mice. The gating strategy to delineate myeloid cell populations is shown in supplemental Figure 5. (A) The absolute number of donor-derived CD45.2+, CD4+ T cells, neutrophils, macrophages, and DCs in the colons of animals 21 days posttransplantation. Results are from a single experiment (n = 3-5/group). (B) The absolute number of donor-derived CD45.2+, CD4+ T cells, neutrophils, macrophages, and DCs in the colons of animals 13 days posttransplantation. (C) The percentage of donor-derived DCs that expressed CD103 and/or CD11b in the colon. Data in panels B and C are from 3 experiments (n = 9-13/group). (D) Representative histograms gated on donor CD64− MHC-II+ CD11c+ cells showing expression of MHC class II, CD80, and CD86 from mice receiving either WT or GM-CSF−/− CD4+ T cells compared with FMO controls. (E) Mean fluorescence intensity of MHC class II, CD80, and CD86 expression on donor CD64− MHC-II+ CD11c+ dendritic cells in replicate mice. Results are from 2 experiments (n = 9-10 per group). (F) mRNA expression of Il23a in sorted donor dendritic cells obtained from pooled colon lamina propria of mice receiving either WT or GM-CSF−/− CD4+ cells on days 13 to 15 posttransplant. Each data point represents pooled results from 5 animals. Data are from 3 independent groups of mice. *P < .05; ****P < .0001.
Donor CD4+ T-cell–derived GM-CSF promotes the activation of colonic dendritic cells. (A-E) Lethally irradiated Balb/c mice were transplanted with B6 Rag-1−/− BM (5 × 106) alone or together with 1 × 106 purified CD4+ T cells from WT or GM-CSF−/− mice. The gating strategy to delineate myeloid cell populations is shown in supplemental Figure 5. (A) The absolute number of donor-derived CD45.2+, CD4+ T cells, neutrophils, macrophages, and DCs in the colons of animals 21 days posttransplantation. Results are from a single experiment (n = 3-5/group). (B) The absolute number of donor-derived CD45.2+, CD4+ T cells, neutrophils, macrophages, and DCs in the colons of animals 13 days posttransplantation. (C) The percentage of donor-derived DCs that expressed CD103 and/or CD11b in the colon. Data in panels B and C are from 3 experiments (n = 9-13/group). (D) Representative histograms gated on donor CD64− MHC-II+ CD11c+ cells showing expression of MHC class II, CD80, and CD86 from mice receiving either WT or GM-CSF−/− CD4+ T cells compared with FMO controls. (E) Mean fluorescence intensity of MHC class II, CD80, and CD86 expression on donor CD64− MHC-II+ CD11c+ dendritic cells in replicate mice. Results are from 2 experiments (n = 9-10 per group). (F) mRNA expression of Il23a in sorted donor dendritic cells obtained from pooled colon lamina propria of mice receiving either WT or GM-CSF−/− CD4+ cells on days 13 to 15 posttransplant. Each data point represents pooled results from 5 animals. Data are from 3 independent groups of mice. *P < .05; ****P < .0001.
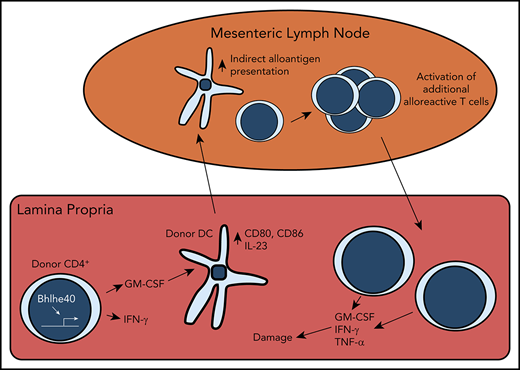
CD4+ T-cell–derived GM-CSF promotes indirect alloantigen presentation during GVHD
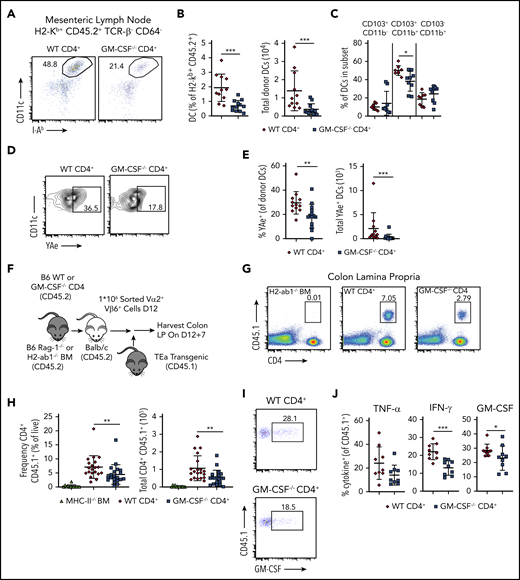
Although recipient hematopoietic and nonhematopoietic APCs are capable of initiating MHC class II-dependent GVHD through the direct pathway,41,42 indirect alloantigen presentation mediated by donor-derived conventional DCs is a critical mechanistic pathway by which this disease is propagated in the GI tract.9,43 Given that CD4+ T-cell–derived GM-CSF enhanced expression of costimulatory molecules on donor-derived DCs, we sought to determine whether this observation was functionally important by examining whether this cytokine facilitated indirect alloantigen presentation. To that end, we observed that animals transplanted with CD4+ T cells from WT mice had a significant increase in the frequency and absolute number of DCs in the mesenteric lymph nodes (mLNs), which has been identified as the primary site for indirect alloantigen presentation9 (Figure 7A-B). In addition, contrary to what was observed in the colon, there was a significant increase in the percentage of CD11b+ CD103+ DC in the mLNs of animals reconstituted with WT grafts (Figure 7C). Using a previously characterized antibody9 that recognizes a Balb/c specific peptide-donor MHC-II complex (YAe), we noted that DCs from the mLNs of mice receiving WT CD4+ T cells presented significantly more host antigen than those from mice receiving GM-CSF−/− CD4+ T cells (Figure 7D-E). To determine whether these disparities in DC number and phenotype affected donor T-cell expansion and inflammatory cytokine production, we employed a methodological approach previously described by Koyama and colleagues9 in which CD4+ T cells were obtained from TEa transgenic mice.44 These cells express a TCR that recognizes a host-specific peptide only when presented on donor MHC class II molecules, thereby providing a readout for indirect presentation. Lethally irradiated Balb/c mice were transplanted with Rag-1−/− BM plus a limiting dose of purified CD4+ T cells from either WT or GM-CSF−/− animals that is not sufficient to cause lethal GVHD. Chimeras were administered sort-purified CD4+ Vα2+ Vβ6+ cells from CD45.1 TEa mice on day 12, and colons were harvested from mice 7 days later (Figure 7F). We observed a significant increase in the frequency and absolute number of TEa T cells in the colons of animals reconstituted with WT vs GM-CSF−/− CD4+ T cells (Figure 7G-H), indicating that CD4+ T-cell–derived GM-CSF promoted indirect alloantigen presentation. There was also an increase in the proportion of TEa T cells that produced IFN-γ and GM-CSF, but not TNF-α (Figure 7I-J). Collectively, these studies demonstrated that the production of GM-CSF by CD4+ T cells augmented indirect alloantigen presentation and promoted the accumulation of proinflammatory CD4+ T cells in the colon.

GM-CSF production by donor-derived CD4+ T cells promotes indirect alloantigen presentation. (A-E). Lethally irradiated Balb/c mice were transplanted with B6 Rag-1−/− BM alone (5 × 106) or together with 1 × 106 purified CD4+ T cells from WT or GM-CSF−/− mice. (A) Representative dot plot gated on donor-derived CD45.2+ TCRβ− CD64− mLN cells 13 days posttransplantation depicting coexpression of CD11c and class II (I-Ab). (B) Frequency and absolute number of dendritic cells in the mLN. Results are from 3 experiments with 11 to 13 mice/group. (C) Percentage of dendritic cells in the mLN that coexpressed CD103 and/or CD11b. Data are derived from 2 experiments with 8 to 9 mice/group. (D-E) Representative flow plots and quantification of the expression of host peptide-donor MHC-II complex (YAe) on donor-derived DCs (CD45.2+ TCRβ−, CD64− CD11c+ MHC-II+). Data are from 3 experiments (n = 11-14/group). (F-J) Lethally irradiated Balb/c mice were transplanted with B6 Rag-1−/− BM (5 × 106) together with 1 × 106 purified CD4+ T cells from WT or GM-CSF−/− mice. Control mice received H2-Ab1−/− (MHC-II-deficient) BM (5 × 106) and 1 × 106 purified CD4+ from WT B6 donors. Sort-purified CD45.1+ CD4+ Vα2+ Vβ6+ cells (1 × 106) from TEa mice were adoptively transferred to mice 12 days posttransplantation. (F) Experimental design. (G) Representative dot plot gated on live, singlet cells from the colons of mice receiving H2-Ab1−/− BM and WT CD4+ T cells or Rag-1−/− BM and either WT or GM-CSF−/− CD4+ T cells 7 days posttransfer. (H) The frequency (expressed as a percentage of total live colon lamina propria cells) and absolute number of CD45.1+ CD4+ TEa cells in the colon 7 days posttransfer. Results are from 3 experiments (n = 9-11 per group). (I) Representative flow plots gated on CD4+ CD45.1+ (ie, TEa) cells depicting GM-CSF production. (J) The percentage of CD45.1+ CD4+ TEa cells in the colon of mice that expressed IFN-γ, GM-CSF, or TNF-α 7 days posttransfer. Results are from 2 experiments (n = 9/group). *P < .05; **P < .01; ***P < .001.
GM-CSF production by donor-derived CD4+ T cells promotes indirect alloantigen presentation. (A-E). Lethally irradiated Balb/c mice were transplanted with B6 Rag-1−/− BM alone (5 × 106) or together with 1 × 106 purified CD4+ T cells from WT or GM-CSF−/− mice. (A) Representative dot plot gated on donor-derived CD45.2+ TCRβ− CD64− mLN cells 13 days posttransplantation depicting coexpression of CD11c and class II (I-Ab). (B) Frequency and absolute number of dendritic cells in the mLN. Results are from 3 experiments with 11 to 13 mice/group. (C) Percentage of dendritic cells in the mLN that coexpressed CD103 and/or CD11b. Data are derived from 2 experiments with 8 to 9 mice/group. (D-E) Representative flow plots and quantification of the expression of host peptide-donor MHC-II complex (YAe) on donor-derived DCs (CD45.2+ TCRβ−, CD64− CD11c+ MHC-II+). Data are from 3 experiments (n = 11-14/group). (F-J) Lethally irradiated Balb/c mice were transplanted with B6 Rag-1−/− BM (5 × 106) together with 1 × 106 purified CD4+ T cells from WT or GM-CSF−/− mice. Control mice received H2-Ab1−/− (MHC-II-deficient) BM (5 × 106) and 1 × 106 purified CD4+ from WT B6 donors. Sort-purified CD45.1+ CD4+ Vα2+ Vβ6+ cells (1 × 106) from TEa mice were adoptively transferred to mice 12 days posttransplantation. (F) Experimental design. (G) Representative dot plot gated on live, singlet cells from the colons of mice receiving H2-Ab1−/− BM and WT CD4+ T cells or Rag-1−/− BM and either WT or GM-CSF−/− CD4+ T cells 7 days posttransfer. (H) The frequency (expressed as a percentage of total live colon lamina propria cells) and absolute number of CD45.1+ CD4+ TEa cells in the colon 7 days posttransfer. Results are from 3 experiments (n = 9-11 per group). (I) Representative flow plots gated on CD4+ CD45.1+ (ie, TEa) cells depicting GM-CSF production. (J) The percentage of CD45.1+ CD4+ TEa cells in the colon of mice that expressed IFN-γ, GM-CSF, or TNF-α 7 days posttransfer. Results are from 2 experiments (n = 9/group). *P < .05; **P < .01; ***P < .001.
Discussion
GVHD within the GI tract is the major determining factor that affects prognosis and overall outcome in patients with this disease.5 Defining the cellular and cytokine networks that initiate and propagate GVHD in this tissue site therefore carries significant clinical implications. To that end, we previously identified a CD4+ CD11c+ IL-23R+ T-cell population that expresses the gut homing molecules CCR9 and α4B7, has a biased central memory phenotype, and plays an important role in initiating early inflammatory events in the colon during GVHD.26 In the current study, we further validated by RNA sequencing analysis that these cells have increased expression of purinergic receptors, complement genes, and Toll-like receptors that confer responsiveness to damage-associated molecular patterns and pattern-associated molecular patterns, which are important in the initiation of GVHD-associated colonic inflammation. Thus, these cells appear to be positioned at the interface of the innate and adaptive arms of the immune system, consistent with a role in mediating early inflammation.
In further characterizing this population, we observed that highly purified CD4+ CD11c+ T cells also have increased expression of Bhlhe40 (also known as Dec1, Stra13, and Sharp2), a transcriptional regulator that was initially described to have a critical role in circadian rhythm.45,46 More recently, Bhlhe40 was found to be expressed in T cells on stimulation through the TCR,47 where it positively regulates GM-CSF22,27 and IFN-γ production,48 a finding we confirmed in mixed lymphocyte cultures. In addition, transplantation of CD4+ T cells from Bhlhe40−/− mice resulted in decreased GVHD-mediated lethality along with a corresponding reduction in pathological damage in the colon. This was accompanied by a reduction in the accumulation of donor-derived CD4+ T cells in the colon, along with decreased numbers of CD4+ GM-CSF+ and CD4+ IFN-γ+ T cells. These data thereby provided confirmation that Bhlhe40+ CD4+ T cells play a critical role in the pathophysiology of GVHD within this tissue site. Because IFN-γ has proven to be a problematic target for the prevention of GVHD due to its differential effects on pulmonary and GI tract inflammation,49 these results led us to focus on the role of GM-CSF in mediating colonic GVHD as a potential therapeutic target.
Although originally characterized as a hematopoietic growth factor, GM-CSF is now well established as a driver of inflammation and serves as a critical link between the adaptive and innate immune systems.11 Although a proinflammatory role for GM-CSF in GVHD has recently been described,20,21 the mechanism by which this cytokine exerts its proinflammatory function has not been delineated. For that reason, a primary aim of this study was to define the mechanistic pathways by which this cytokine induced pathology in the GI tract. Prior reports have shown that inflammatory cytokines can perpetuate inflammation in the colon by inhibiting the reconstitution of Tregs.28,50 Therefore, it was possible that a mechanism by which CD4+ T cell production of GM-CSF exacerbated GVHD was by inhibiting the effective restoration of the regulatory T-cell compartment. To that end, several studies have demonstrated that exogenous GM-CSF administration induces a tolerogenic DC population that effects the expansion of CD4+ Foxp3+ Tregs, which are then able to suppress autoimmunity.51-53 Contrary to these studies, however, we observed that GM-CSF had no discernible effect on Tregs, as neither induced nor natural Treg reconstitution was adversely affected. Moreover, IL-10 expression, which is a major suppressive pathway by which Tregs inhibit GVHD severity,37 did not differ in Tregs from animals transplanted with WT vs GM-CSF−/− marrow grafts. Thus, our results do not support a deleterious role for GM-CSF in the quantitative or qualitative alteration of Treg function within the GI tract.
We therefore focused on how GM-CSF modulated the effector, as opposed to the regulatory, arm of the immune system. Because T cells do not express the GM-CSF receptor,19 the inflammatory effects of this cytokine are presumably mediated through myeloid cell populations. In fact, the GM-CSF-induced accumulation of dendritic cells, neutrophils, and monocytes in organs such as the spleen and liver has been reported in GVHD.20 However, the precise mechanism by which these cells promote inflammation, particularly in the GI tract, has not been defined. In the present study, we observed that the accumulation of WT CD4+ T cells in the GI tract leads to increased expression of costimulatory molecules (ie, CD80 and CD86) and IL-23 in donor-derived conventional DCs when compared with CD4+ T cells that lack GM-CSF. This is consistent with studies that have demonstrated that GM-CSF is important for the maturation of tissue-resident DC populations12,54 and can stimulate IL-23 production from DCs.39,55 CD4+ T-cell–derived GM-CSF production also increased the number of DCs in the mLNs, which have been shown to be the primary site for indirect alloantigen presentation during GVHD.9 In addition, we observed that there were increased numbers of alloreactive T cells that had a proinflammatory cytokine signature, indicating that one of the key roles of GM-CSF is indeed to promote indirect alloantigen presentation. Interestingly, we observed an increased frequency of CD103+ CD11b+ DCs in the mLN, which is a subset has been shown to arise from DC-restricted progenitors, be regulated by GM-CSF, produce IL-23, and have a superior ability to traffic to the MLN.12,56,57 CD103+ DCs that enhance alloantigen presentation in the colon have been previously identified by Koyama and colleagues,9 although the relevant CD103+ DC population in their studies lacked expression of CD11b. This phenotypic difference may be, in part, because animals in our model were transplanted with purified CD4+ T cells, which are primary producers of GM-CSF and therefore perhaps more likely to drive the accumulation of a GM-CSF-responsive CD103+ CD11b+ DC population than animals reconstituted with unseparated T cells.
Prior preclinical studies have demonstrated that IL-6 and IL-23 play important roles in mediating GVHD-induced damage in the GI tract.29-32 In addition, clinical trials have shown that blockade of IL-6, specifically, can provide protection from pathological damage in this tissue site.58,59 IL-6 and IL-23 have also been shown to stimulate production of GM-CSF in murine models of neuroinflammation,39,60 suggesting they could function as upstream regulators of this cytokine. However, we observed that antibody-mediated inhibition of either IL-6 or IL-23 had no effect on the frequency or absolute number of CD4+ GM-CSF+ T cells in the colon. These results indicated that neither cytokine appeared to be important for lineage differentiation, which is consistent with a prior report supporting a role for IL-7 in this process.21 These data also support the premise that GM-CSF constitutes a nonredundant inflammatory pathway within the GI tract independent of other cytokine mediators such as IL-6 and IL-23. The implication of these results from a clinical perspective is that inhibition of GM-CSF might provide additional protection when coupled with strategies designed to block these other cytokines. To that end, mavrilimumab,61 an anti-GM-CSF receptor α monoclonal antibody, and MOR103,62 a humanized anti-GM-CSF antibody, are currently in clinical trials for the treatment of rheumatoid arthritis and multiple sclerosis, respectively. Mavrilimumab was found to significantly decrease disease activity in a phase 2 study in patients with rheumatoid arthritis,61 raising the possibility that this agent might ultimately be available for off-label use in GVHD for the prevention of inflammation and pathological damage in the GI tract. Furthermore, GM-CSF is rarely used as a growth factor in recipients of allogeneic hematopoietic stem cell transplantation, and has been shown to exacerbate GVHD in murine models,21 so therapies targeting endogenous GM-CSF production would appear to be a clinically viable approach.
In summary, we have identified a pathogenic Bhlhe40+ GM-CSF+ CD4+ T-cell population that drives lethal gastrointestinal damage during acute GVHD. Our studies support a model whereby production of GM-CSF by alloreactive CD4+ T cells, which is transcriptionally regulated by Bhlhe40, enhances the activation of donor-derived APCs in the colon, which subsequently traffic to the mLN, where they can promote indirect alloantigen presentation. This leads to the activation and expansion of proinflammatory donor T cells, which emigrate to the colon, where they mediate pathological damage. Thus, GM-CSF serves as a critical link between the adaptive and innate arms of the immune system, positioning this cytokine as a key regulator of GVHD in the colon and a potential therapeutic target for amelioration of this disease.
For original data, please contact the corresponding author.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This research was supported by National Institutes of Health (NIH), National Heart, Lung, and Blood Institute grants F30 HL143870 (C.P.), R01 HL064603 (W.R.D.), R01 HL126166 (W.R.D), R01 HL139730 (J.S), NIH National Cancer Institute grants R01 CA163725 (J.S) and K12 CA120780 (B.V.), and NIH, National Institute of Allergy and Infectious Diseases grant R01 AI113118 (B.T.E). B.V. is also supported by a grant from the University of North Carolina University Cancer Research Fund. C.P. is a member of the Medical Scientist Training Program at the Medical College of Wisconsin, which is partially supported by a training grant from the National Institute of General Medical Sciences T32-GM080202.
Authorship
Contribution: C.P. conducted and designed experiments and wrote the manuscript; V.Z. performed experiments; R.K. performed blinded pathological analysis; A.S. performed biostatistical analysis; B.V. and J.S. assisted with RNA sequence analysis and edited the manuscript; M.-L.A., B.T.E., and R.T. provided critical reagents and edited the manuscript; and W.R.D. designed experiments, supervised the study, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: William R. Drobyski, Bone Marrow Transplant Program. 8701 Watertown Plank Rd, Milwaukee, WI 53226; e-mail: wdrobysk@mcw.edu.