

In this issue of Blood, report that hepcidin secreted by the fetal liver has a specific role in iron homeostasis, ensuring that the fetal liver retains iron destined for hepatic erythropoiesis.1 Surprisingly, fetal hepcidin seems to have no physiological role in regulating iron transfer from the mother to the fetus across the placenta. Why this is so is the subtext of this article and the focus of other recent analyses.2
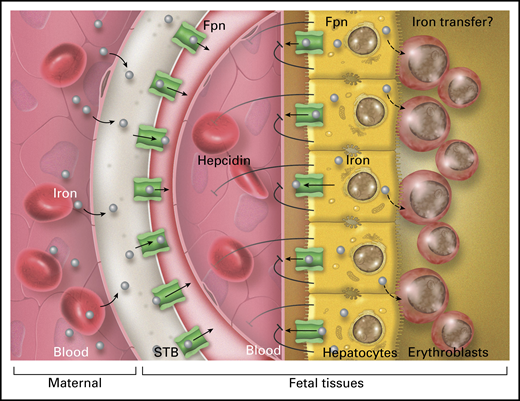
Hepcidin secreted by the fetal liver does not appreciably affect placental iron delivery to the fetus through ferroportin (Fpn) on syncytiotrophoblast (STB) but does inhibit the efflux of iron from fetal hepatocytes through Fpn into fetal blood, presumably by an autocrine/paracrine mechanism. Retention of iron in fetal hepatocytes is required for effective fetal erythropoiesis in the liver, possibly because hepatocytes can transfer iron to adjacent erythroblasts. Professional illustration by Somersault18:24.
Hepcidin secreted by the fetal liver does not appreciably affect placental iron delivery to the fetus through ferroportin (Fpn) on syncytiotrophoblast (STB) but does inhibit the efflux of iron from fetal hepatocytes through Fpn into fetal blood, presumably by an autocrine/paracrine mechanism. Retention of iron in fetal hepatocytes is required for effective fetal erythropoiesis in the liver, possibly because hepatocytes can transfer iron to adjacent erythroblasts. Professional illustration by Somersault18:24.
Normal human pregnancy challenges the mother with a greatly increased demand for iron to support placental and fetal growth, including importantly that of the developing fetal erythron, but additional iron is also needed for expanding maternal erythropoiesis. The net effect is an ∼10-fold increase in iron demand from 0.8 mg/day in the first trimester to 7.5 mg/day in the third trimester. In the second and third trimesters, the maternal iron-homeostatic system responds to this challenge by gradually decreasing the production of maternal hepcidin in the liver, resulting in very low maternal circulating hepcidin concentrations.2 Low hepcidin concentrations allow greatly increased intestinal iron absorption when dietary iron is available and mobilization of iron from maternal stores. What causes maternal hepcidin suppression is not yet known.
The syncytiotrophoblast is the placental tissue that carries out nutrient transport from maternal blood to fetal blood and the removal of fetal waste in the opposite direction (see figure). In humans, this is a single polarized cell layer; in mice, there are 2 cellular layers that seem to be interconnected so that they function as a single layer. In both species, the syncytiotrophoblast functionally separates the maternal from the fetal milieu. Iron uptake on the maternal side is mediated by the transferrin receptor TFR1, and the iron is then exported to the fetal vasculature through ferroportin, the sole known cellular iron exporter and the molecular target of hepcidin. Hepcidin, if present at effective concentrations, regulates iron export through ferroportin by occluding this transporter and inducing its endocytosis and lysosomal proteolysis. Because placental ferroportin is localized on the fetal-facing side of the syncytiotrophoblast tissue, only fetal hepcidin has direct access to it.
Kämmerer et al used several mouse models to examine fetal iron homeostasis. In the first model, they studied wild-type fetuses of wild-type mothers, and found that as iron accumulated in the fetal liver, its hepcidin messenger RNA (mRNA) concentration increased but remained well below the already low maternal liver hepcidin mRNA concentrations, although hepcidin peptide in plasma was not measured. Nevertheless, fetal hepcidin concentrations are likely too low to affect placental ferroportin because placental ferroportin was comparable between littermate fetuses lacking hepcidin or not.2 In the study by Kämmerer et al, even when the pregnant mouse received an iron overload diet, and fetal liver hepcidin increased in response, placental ferroportin protein did not decrease, indicating that fetal hepcidin activity was insufficient to exert its expected suppressive effect on placental ferroportin. In this setting, ferroportin is predominantly regulated by the cellular homeostatic effect of the iron regulatory element-iron regulatory protein system.2 Yet ferroportin on the fetal surface of the syncytiotrophoblast is capable of responding to substantially higher concentrations of fetal hepcidin, as shown by mouse fetuses with ablated Tmprss6genes or with transgenic fetal hepcidin overexpression. In both of these models, high fetal hepcidin concentrations caused fetal iron deficiency and anemia.3 Fetal iron restriction may also develop with high fetal hepcidin concentrations generated during fetal infection or inflammation.4
To determine whether the fetal hepcidin-ferroportin interaction has any baseline function, Kämmerer et al generated fetuses with disabled fetal hepcidin activity either by knocking in a hepcidin-unresponsive mutant of ferroportin C326Y in all fetal tissues or only in hepatocytes, or by ablating the hepcidin gene in fetal hepatocytes. The fetal phenotypes differed in subtle detail, but they shared a small but reproducible decrease of iron retention in the liver and transient anemia detected 2 days before term but not at term. This is indicative of an autocrine/paracrine effect of hepcidin secreted by fetal hepatocytes and acting on ferroportin in the same cells to prevent iron export from these cells into fetal circulation. A similarly unexpected autocrine/paracrine effect of hepcidin on another cell type, cardiomyocytes, was previously demonstrated by Kämmerer et al.5 The transient fetal anemia suggests that the retained hepatocyte iron directly supports erythropoiesis in the liver but how this occurs is not yet known. By the time the mouse pups are born, erythropoiesis has transitioned largely to their marrow, and the autocrine effect of hepcidin on hepatocytes no longer matters for erythropoiesis.
Importantly, there was no detectable effect of these manipulations on the placental iron content, transporter expression, or ferroportin protein, confirming that physiologic fetal hepcidin concentrations are too low to exert an endocrine hormonal effect in the fetus.
Finally, the article by Kämmerer et al and other recent data2 indicate that during maternal iron deficiency, the fetus is not treated preferentially and also becomes iron deficient. As shown by others,2 the organ that seems to be protected is the placenta, perhaps because it requires iron for mitochondria to maintain the energy required for all nutrient and waste transport. The lack of effective fetal compensation for maternal iron deficiency during pregnancy may explain why maternal iron deficiency remains an important global health problem with long-term consequences for children.
Conflict-of-interest disclosure: The author declares no competing financial interests

