Abstract
Relapsed acute lymphoblastic leukemia (ALL) has remained challenging to treat in children, with survival rates lagging well behind those observed at initial diagnosis. Although there have been some improvements in outcomes over the past few decades, only ∼50% of children with first relapse of ALL survive long term, and outcomes are much worse with second or later relapses. Recurrences that occur within 3 years of diagnosis and any T-ALL relapses are particularly difficult to salvage. Until recently, treatment options were limited to intensive cytotoxic chemotherapy with or without site-directed radiotherapy and allogeneic hematopoietic stem cell transplantation (HSCT). In the past decade, several promising immunotherapeutics have been developed, changing the treatment landscape for children with relapsed ALL. Current research in this field is focusing on how to best incorporate immunotherapeutics into salvage regimens and investigate long-term survival and side effects, and when these might replace HSCT. As more knowledge is gained about the biology of relapse through comprehensive genomic profiling, incorporation of molecularly targeted therapies is another area of active investigation. These advances in treatment offer real promise for less toxic and more effective therapy for children with relapsed ALL, and we present several cases highlighting contemporary treatment decision-making.
Introduction
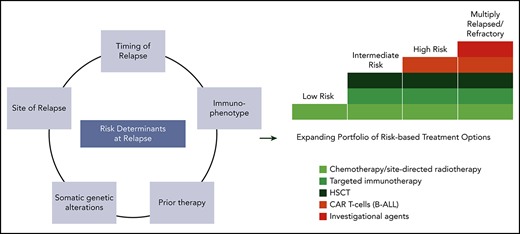
More than 85% of children with acute lymphoblastic leukemia (ALL) survive without relapse following contemporary therapies,1,2 but survival following relapse is poor. Among 1961 children who enrolled in Children’s Cancer Group ALL trials between 1988 and 2002 and relapsed, the 5-year overall survival (OS) rate was 36%.3 Similarly 10-year OS was 36% for children treated in the Acute Lymphoblastic Leukemia Relapse Berlin-Frankfurt-Münster (ALL-REZ-BFM) 90 trial.4 Contemporary data show 5-year OS rates of ∼50% for first relapse of ALL.5,6 Risk factors for outcome following first relapse have been defined and incorporated into risk stratification schemes: time from diagnosis to relapse (shorter is worse), site of relapse (marrow worse than extramedullary), immunophenotype (T worse than B), and minimal residual disease (MRD) response to reinduction therapy (Table 1). Survival following second or later relapses or relapses after hematopoietic stem cell transplantation (HSCT) has historically been much worse, although newer immunotherapies such as chimeric antigen receptor-redirected (CAR) T cells may change this.7 Critical questions to consider include which chemotherapy regimen to use for relapsed ALL, when HSCT should be used in second complete remission (CR2), and the role of immunotherapies including blinatumomab, inotuzumab ozogamicin, and CAR-T cells. Using illustrative cases, we describe our approach to these challenges, which is informed by our experience with Children’s Oncology Group (COG) trials.8-12
Patient 1: first bone marrow relapse of B-ALL
A 12-year-old girl diagnosed with B-ALL 2 years ago has an isolated marrow relapse during maintenance therapy. What reinduction should be used and what postinduction therapy is optimal? Should she undergo HSCT in CR2? What if the relapse occurred after completion of therapy, 40 months following diagnosis? Should somatic genetic alterations alter the approach?
Discussion and proposed treatment
Relapsed ALL therapies differ significantly between cooperative groups, but attain similar outcomes (Table 2).10,11,13-16 We would administer the United Kingdom (UKALL) R3 reinduction regimen (dexamethasone, vincristine, mitoxantrone, pegaspargase, and intrathecal methotrexate)15 used in the recent COG AALL1331 first relapse B-ALL trial (NCT02101853) or another 4-drug reinduction regimen. Because infectious risk is high,17,18 we would hospitalize her until count recovery and provide antibacterial, antifungal, and Pneumocystis jirovecii prophylaxis.19,20 If she has an M3 marrow (>25% marrow blasts) response to reinduction, we would use the CD22 antibody-drug conjugate inotuzumab and/or CAR-T cell therapy (see the following section).7,21,22 If she enters CR2 or has an M2 marrow (5%-25% blasts), we would administer 2 28-day cycles of blinatumomab, followed by HSCT in CR2 if she is MRD− (Figure 1A). If a donor is readily available, we would consider HSCT after cycle 1 if she is MRD−. Blinatumomab, a bispecific T-cell-engaging antibody that links CD3+ T cells to CD19+ B-ALL cells, is active in relapsed and refractory (r/r) adult and pediatric ALL.23,24 COG AALL1331, which compared 2 cycles of UKALL R3 postinduction chemotherapy to 2 cycles of blinatumomab, was recently stopped early because of improved disease-free survival, superior OS, lower toxicity, and superior MRD clearance with blinatumomab.17 Survival following HSCT is similar with HLA-matched siblings, fully matched unrelated donors, and haploidentical donors, so we would pursue HSCT using the best available donor.25,26 Because patients that remain MRD+ before HSCT have inferior outcomes, we would consider other treatment strategies if MRD was ≥0.01% following 2 blinatumomab cycles.27 Inotuzumab is 1 option, although it may increase the risk of veno-occlusive disease post-HSCT, particularly if multiple cycles are administered.21,22 Newer next-generation sequencing-based MRD detection methods may allow more precise risk classification,28 but have not been generally incorporated into decisions regarding HSCT in CR2. Clinical trials are testing CAR-T cells in patients with high-risk first relapse (NCT04276870, NCT02443831) and it would be reasonable to consider enrollment in a trial, particularly with a poor response to reinduction. In that case, we would be cautious with the use of blinatumomab before planned CAR T-cell therapy (see the following section).
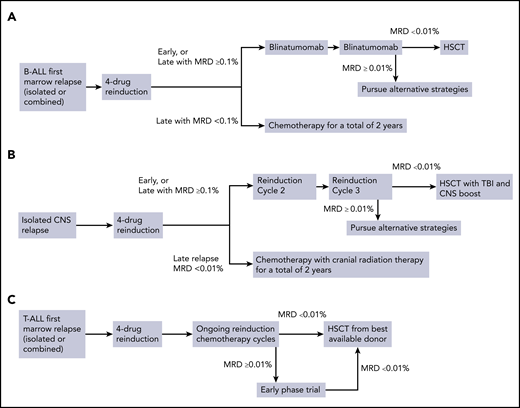
Treatment algorithms for children with relapsed ALL. (A) Treatment of marrow relapses of B-ALL. (C) Treatment of isolated CNS relapse. (C) Treatment of relapsed T-ALL.
Treatment algorithms for children with relapsed ALL. (A) Treatment of marrow relapses of B-ALL. (C) Treatment of isolated CNS relapse. (C) Treatment of relapsed T-ALL.
Time from diagnosis to relapse is highly prognostic. Exact times and terminologies differ between groups, but <18 months is generally considered very early, 18 months to 3 years (or the end of chemotherapy for the BFM) is early, and >3 years (or after therapy completion) is late.6,14,15,17 HSCT is often used for patients with late marrow relapse and a poor MRD response after the first reinduction cycle, with chemotherapy alone used for good responders. Most groups use MRD ≥0.1% as the HSCT threshold, but some use MRD ≥0.01%.10 For patients with late marrow relapse and a good MRD response, the BFM, COG, and UKALL trials have observed long-term OS ∼70% with different multiagent chemotherapy regimens.10,13,29 If she had late marrow relapse with MRD <0.1% at end reinduction, we would use the UKALL R3 regimen because it has lower cumulative doses of chemotherapy, but others might chose the ALL-REZ-BFM 2002 or COG AALL0433 regimens. If her MRD was ≥0.1%, we would treat her as outlined previously with 1 to 2 cycles of blinatumomab followed by HSCT (Figure 1A).
Although somatic genetic alterations are not routinely used in risk assignment following relapse, several unfavorable alterations have been identified and could influence treatment decision-making. We would perform panel sequencing and RNA-based fusion gene testing.30,31 If the Philadelphia chromosome were present, we would add imatinib or dasatinib to reinduction therapy and would strongly consider use of these agents if an ABL-class gene fusion was detected.32 One might also consider addition of ruxolitinib if a JAK2 fusion were detected, but the efficacy and safety of this strategy have not yet been proven, so we would use ruxolitinib only if she were refractory to reinduction or remained MRD+ after 2 cycles of blinatumomab. TP53 mutations can be acquired at relapse and are associated with very poor outcome. If present, it would be reasonable to consider HSCT or CAR-T cells in CR2 regardless of time to relapse or MRD response.13,33,34 If she had a late marrow relapse with a high-risk somatic genetic alteration, such as TCF3-HLF fusion, we would consider HSCT or CAR-T cells in CR2 regardless of MRD response.
Patient 1, scenario 2: relapse following HSCT
Patient 1 was MRD− following reinduction, received 2 blinatumomab cycles, and then matched sibling donor HSCT. She had no graft-versus-host disease and immunosuppression was stopped 6 months post-HSCT. Thirteen months post-HSCT, she had a second marrow relapse with 80% blasts and 98% donor T cells in peripheral blood. What therapy should she receive next? What should be used for definitive therapy?
Discussion and proposed treatment
Until recently, children and adolescents with ALL that relapsed post-HSCT had a dismal outcome, with multiple complications associated with second allogeneic transplants and long-term survival rates of only 25% to 30% if remission is achieved.35-38 The time between HSCT and relapse is prognostic, with low survival for relapse within 6 to 12 months, but better outcomes for those that relapse >12 months post-HSCT and undergo second transplant in remission.38
Adoptive cell therapy has revolutionized the treatment of relapsed ALL, creating new opportunities for long-term survival. The early efficacy of CD19-redirected CAR T-cells in r/r ALL is well-established7,39,40 ; a complete discussion is beyond the scope of this review (see Aldoss and Forman41 ). Currently, tisagenlecleucel is the only US Food and Drug Administration-approved CAR-T product for pediatric ALL (also approved by the European Medicines Agency and other regulators); more products will likely be approved in the next 1 to 2 years. This therapy, termed CTL019 during its early clinical development at the University of Pennsylvania and the Children’s Hospital of Philadelphia (CHOP), was shown to be active in single-center trials leading to the definitive phase 2 registration trial (termed ELIANA) conducted at 25 sites worldwide.7,42,43 Seventy-five r/r ALL patients 3 to 21 years old were infused with CTL019, with >95% first receiving lymphodepleting chemotherapy.7 In the initial ELIANA report, 81% of patients attained an MRD− complete response (CR) within 3 months and 12-month event-free survival (EFS)/OS was 50%/76%. The patients were heavily pretreated (median 3 prior therapies, 61% had prior HSCT). Much more data are needed to define the long-term efficacy of tisagenlecleucel and other CAR-T cell therapies for r/r ALL, but the first patient treated at CHOP in 2012 continues to do very well, with no additional antileukemia therapy given, 8 years postinfusion,42 and more mature ELIANA data show a 2-year recurrence-free survival rate of 62% among those achieving remission.44
For patient 1, given the limitations of second HSCT, our goal would be to administer tisagenlecleucel or to enroll in a clinical trial testing other CAR-T cell therapies. Ideally, we would collect T cells before administering any antileukemic therapy. This is generally feasible, even with overt relapse, if the peripheral blood T-cell count is >200/μL. The risk of graft-versus-host disease following CAR T-cell therapy after allogeneic HSCT is quite low, and symptoms are generally mild.45 If the T-cell count is too low or T-cell collection is not feasible, we would administer chemotherapy to debulk leukemia burden, hoping to avoid infections or other complications because it is not essential for the patient to be in CR for CAR-T cells to be effective and serious infections or end-organ toxicity can complicate subsequent CAR T-cell therapy. We often use a simple 3-drug induction with corticosteroids, vincristine, and pegaspargase, a combination such as cyclophosphamide and etoposide, or a course of high-dose methotrexate for these purposes.45 If the patient receives chemotherapy, then she will need to be at least 2 weeks postchemotherapy and have an absolute lymphocyte count >500/μL before collection. Although inotuzumab is very active in r/r ALL (see the following section),22 we would avoid it if possible in this setting as it can produce long-term B-cell aplasia that might affect CAR-T cell expansion postinfusion by reducing the number of CD19+ target cells that prime expansion.45 However, there is no contraindication to inotuzumab before planned CAR-T cell therapy and we have used it in many patients.
Following collection, CAR-T cell manufacture takes about 4 weeks. We would continue with low-intensity chemotherapy until 2 weeks before the planned infusion date.45 One week preinfusion, we would administer lymphodepleting chemotherapy (fludarabine 30 mg/m2 days 1-4 and cyclophosphamide 500 mg/m2 days 1-2) followed by CAR-T cell infusion. As long as she is healthy, outpatient management is feasible, though many are admitted for fever. We would monitor closely for cytokine release syndrome, which can be treated with tocilizumab, a monoclonal antibody directed against the interleukin-6 receptor.45,46
A key question for r/r ALL is whether CAR-T cells should be a definitive therapy or a “bridge” to HSCT. This is an area of significant unknowns, which have been addressed recently.41,45 Many patients remain in long-term remission without further therapy, but challenges remain in selecting which patients should undergo HSCT post-CAR; decisions may depend on the specific CAR-T cell product, because CAR-T cell persistence is influenced by the nature of the CAR costimulatory domain. Like this patient, most children treated with CAR-T cells have relapsed following allogeneic HSCT, and avoiding a second HSCT is attractive. Key factors to consider include the length (and perhaps depth) of MRD response and the duration of B-cell depletion, a useful biological surrogate for activity because CAR T-cells also kill normal B cells. Patients with early MRD recurrence or loss of B-cell depletion probably will not be cured with CAR-T cells alone and require HSCT. The exact length of B-cell depletion required is unknown, but is likely at least 6 to 12 months. Notably, only 9% of ELIANA trial patients underwent subsequent HSCT; all 8 were alive at last report.7,45 We would monitor this patient with bone marrow aspirate/biopsy and MRD testing at days 30 and 90 and every 3 months for the first year and quantitate peripheral blood B cells monthly for at least the first 6 months postinfusion. We would consider any evidence of MRD recurrence by conventional measures (flow cytometry or immunoglobulin/T-cell receptor polymerase chain reaction) or B-cell recovery within the first 6 months to be an indication for a second HSCT. If the patient remained MRD− and without B-cell recovery for at least 6 months, then we would continue to monitor closely without additional therapy, gradually reducing the testing frequency. Emerging data suggest that more sensitive next-generation sequencing MRD testing may help determine who needs HSCT after CAR-T cell therapy.47,48
When ALL relapses post-CAR, it can be CD19+ or CD19− (antigen escape). Mechanisms of escape include point mutations, frameshift mutations, and alternative splicing of CD19 transcripts, removing the domain recognized by the CAR CD19 antibody.49,50 A potentially important issue in this patient is her prior blinatumomab therapy. Although data are not conclusive, prior blinatumomab therapy may increase the risk of failure to attain an MRD− response or CD19− relapse post-CAR.51 We would not use the prior blinatumomab therapy to decide on a second HSCT post-CAR, but would monitor her closely.
Patient 1, scenario 3: relapse following CAR T-cell therapy
She attained an MRD− CR after tisagenlecleucel and remained MRD− and with B-cell depletion for 13 months, when she was found to have a CD19−, but CD22+ isolated marrow relapse. Do potentially curative options exist? What treatment should be considered? Would options be different if the relapse were CD19+?
Discussion and proposed treatment
CAR-T cells are not a panacea; most trials show that 25% to 50% of patients with a CR will subsequently relapse, with longer follow-up needed to refine relapse risk.52 We believe that patient 1 still has some chance for cure, but would have a detailed discussion with her and her family about therapeutic options, including palliative therapy.
Because the relapse is CD19−, blinatumomab and CD19-CAR are not options. She has received very little chemotherapy since her initial relapse, so one can consider some of the options discussed for patient 3. She is now about 2.5 years post-HSCT and in good clinical condition, so a second allogeneic HSCT is feasible if she has an MRD− response to therapy and remains in good medical condition. There are also clinical trials testing CD22-redirected CAR T-cells (NCT02315612, NCT04088864, NCT02650414). Our early and limited experience with CD22-CAR suggests that MRD− responses are common, but that they are less durable than those with CD19-CAR. We believe that this teenager would likely need to undergo a second HSCT to be cured; hence, it will be critical to minimize toxicity. We would administer inotuzumab or enroll in a trial of CD22-CAR with the goal of attaining an MRD− CR (her fourth). If CD22-CAR were pursued, then low-intensity chemotherapy could be administered while awaiting collection/manufacture. Inotuzumab is immediately available, but may also increase the risk of veno-occlusive disease after a second transplant.21 In this setting, all approaches have risks, so we would carefully consider preexisting toxicities and patient and parent preferences.
If this relapse were still CD19+, then other options exist, including treatment with blinatumomab or retreatment with CD19-CAR, expanding the therapeutic options discussed previously. The patient could be re-treated with tisagenlecleucel or could enroll in a clinical trial testing other CAR strategies such as CARs targeting both CD19 and CD22 (NCT03448393, NCT03241940) or humanized CD19-CAR (NCT02374333). Early data from the CHOP humanized CTL119 CD19-CAR trial show that 9/16 (56%) patients previously treated with CTL019 achieved CR with B-cell aplasia, which was MRD− in 7/9 responders, with 1-year recurrence-free survival rate 56% among responders.53 If retreatment were pursued, then detailed discussions should occur regarding whether to pursue a second HSCT if MRD− CR4 were achieved.
Patient 1, scenario 4: multiply relapsed ALL
Patient 1 received CD22-redirected CAR-T cells on a clinical trial, and attained an MRD− CR. However, she had another marrow relapse while awaiting a second HSCT. Are potentially curative options still available? What treatment could be considered?
Discussion and proposed treatment
Multiply relapsed ALL after HSCT and CAR-T cell therapy is very challenging to treat. Potentially curative options may exist, assuming that she is healthy, as she has not undergone a second HSCT, but this is also a situation in which focus could be directed at maximizing quality of life, rather than cure. Those issues should be explored thoroughly with the patient and her family. Even if not a pathway to cure, remission could improve quality of life.
If the blasts remain CD22+, then we would use inotuzumab either as a single agent54 or in combination with chemotherapy (ITCC-059; EudraCT 2016-000227-71). One could also consider investigational clinical trials or palliative therapy (Table 3). If not done previously, we would perform comprehensive genomic profiling to assess for potentially targetable alterations to inform matched targeted therapy (NCT02670525).55 Alterations in many biological pathways have been implicated in r/r B- and T-ALL, and treatment with an mTOR inhibitor (NCT01523977; NCT01614197, NCT03328104, NCT01614197), proteasome inhibitor (NCT02303821, NCT03888534, NCT03817320), CDK4/6 inhibitor (NCT03792256, NCT03515200), or BCL-2 inhibitor (NCT03236857) in combination with chemotherapy could be considered (Table 3). Optimization of traditional agents could also be considered. For example, a phase 1 trial investigating liposomal vincristine in children with refractory leukemia was completed that established safety and preliminary single agent activity.56 Liposomal vincristine is being investigated in combination with the UK ALLR3 regimen (NCT02879643) in pediatric patients with r/r ALL and this regimen could be considered, weighing the risks and benefits of intensive cytotoxic therapy in this setting.
Patient 2: first extramedullary relapse of B-ALL
A 6-year-old boy diagnosed with B-ALL 15 months ago has an asymptomatic isolated central nervous system (iCNS) relapse during maintenance therapy, with 10 white blood cells/μL cerebrospinal fluid with blasts on cytospin, and an MRD− bone marrow. What reinduction regimen should be used? If he enters remission, what postinduction therapy is optimal? Should he undergo HSCT in CR2? Would he be managed differently if the relapse occurred 30 months following diagnosis? Would marrow MRD impact management? Is there a role for CAR-T cells?
Discussion and proposed treatment
Because most subsequent treatment failures following iCNS relapse involve the marrow, intensive reinduction strategies are used plus CNS-directed therapy, classically cranial irradiation.10,57,58 Although outcomes for iCNS relapses are better than marrow or combined relapses with 5-year OS 65%,6 early iCNS relapses (<18 months from diagnosis) have inferior outcomes with 3-year EFS/OS rates of 41%/52% on COG AALL0433 and similar outcomes from other groups.10 Randomized trials comparing HSCT to chemotherapy in this population have not been feasible, but because survival is only ∼50%, many groups treat early iCNS relapses with CR2 HSCT. With small patients numbers, nonsignificant trends favoring HSCT over chemotherapy and cranial radiation were reported on COG AALL0433, UKALL R3, and single institution studies.10,59,60 Retrospective analysis of Italian children treated with HSCT for isolated extramedullary relapse from 1990 to 2010 showed improvements in 10-year survival rates for very early isolated extramedullary relapses <18 months from diagnosis from historical rates of 20% to 30% with chemo/radiotherapy to 52% with HSCT.61
We would treat patient 2 with the dexamethasone-based UKALL R3 reinduction with weekly triple intrathecal chemotherapy (Figure 1B). Triple intrathecal chemotherapy is generally considered superior to intrathecal methotrexate alone for patients with overt CNS leukemia.58 Omaya reservoir placement can be considered for frequent delivery of intrathecal chemotherapy. Following attainment of remission with 3 blocks of intensive systemic reinduction therapy, we would recommend best available donor HSCT following a total body irradiation preparative regimen with a CNS boost.
Time to relapse is an important prognostic factor in extramedullary relapse. The EFS rates for children with late iCNS relapses (≥18 months from diagnosis) are 75% to 80% with chemotherapy with both systemic and CNS effects (eg, intermediate or high-dose methotrexate and cytarabine) and delayed CNS irradiation to maximize early delivery of intensive chemotherapy.62 Had patient 2 relapsed 30 months postdiagnosis, we would recommend intensive chemotherapy for 2 years with delayed cranial radiation (1800 cGy) around 12 months.59 Many patients with isolated extramedullary relapses have submicroscopic marrow MRD at the time of relapse.63,64 Given the adverse prognostic significance of persistent MRD at the end of reinduction,65,66 we would only recommend HSCT if he remained marrow MRD+ at the end of reinduction, which is very uncommon.
Efforts are under way to reduce the acute and long-term toxicities associated with intensive chemotherapy and cranial radiation. Although an increase in treatment failures was observed on the COG AALL02P2 trial, where the dose of cranial radiation therapy was further reduced to 1200 cGy,67 alternative therapies are being investigated. CAR-T cells circulate in the cerebrospinal fluid,43 and clinical trials testing this modality could be considered (NCT04276870), especially if the initial response to reinduction is suboptimal, HSCT is contraindicated, or the patient is very young with increased risk of long-term toxicity following cranial radiotherapy.68
Patient 3: first marrow relapse of T-ALL
An 8-year-old boy with T-ALL has an isolated marrow relapse 17 months postdiagnosis. Initial therapy included a dexamethasone-based 4-drug induction followed by augmented BFM-based chemotherapy with nelarabine with intrathecal chemotherapy for CNS prophylaxis.69 What reinduction regimen should be used? If he enters CR2, should he undergo HSCT? What are the treatment options if he does not respond to salvage therapy? Is there a role for immunotherapy?
Discussion and proposed treatment
Relapses occur earlier for T-ALL than B-ALL, with most happening within 2 years of diagnosis.70 Survival postrelapse is poor with OS rates of <25% with marginal recent improvements.6,71,72 Given these outcomes, HSCT is the preferred option for relapsed T-ALL, regardless of the site/timing of relapse. We recommend reinduction chemotherapy followed by HSCT with the best available donor as soon as an MRD− CR is achieved (Figure 1C). There is no standard reinduction approach for T-ALL or established superiority of 1 regimen over another. As for patient 1, we would use the UKALL R3 reinduction regimen, with hospitalization until count recovery and antimicrobial prophylaxis.15 Although only a small number of children with T-ALL were enrolled on this trial, 3-year progression-free survival rates on the mitoxantrone arm were 65%. Another potential reinduction regimen is nelarabine, cyclophosphamide, and etoposide (NECTAR) that showed CR2 rates of 44% in a phase 1 trial,73 and 5/5 patients with r/r T-ALL achieved CR in another series, with acceptable neurotoxicity.74 Concomitant administration of nelarabine and intrathecal chemotherapy should be avoided to reduce the risk of neurotoxicity, so NECTAR may be more practical to administer postinduction, especially if CNS involvement is present. The COG AALL07P1 regimen (bortezomib added to a prednisone-based 4-drug reinduction), that had CR2 rates of 68 ± 10% in 22 T-ALL patients, could also be considered.9
Although somatic genetic alterations are not routinely used for risk-based treatment allocation in newly diagnosed/relapsed T-ALL, we recommend comprehensive molecular profiling at relapse, if available. Targetable ABL-class fusions have been reported and JAK-STAT pathway activation has been observed in early T-cell precursor ALL.75-77
There is growing enthusiasm for immunotherapy approaches in r/r T-ALL.78 The CD38-directed monoclonal antibody daratumumab has shown promising preclinical activity in T-ALL and early T-cell precursor ALL,79 and daratumumab combined with chemotherapy is being tested in r/r T-ALL (NCT03384654).
A number of biological pathways have been implicated in T-ALL and several regimens have or are currently investigating molecularly targeted agents in T-ALL relapse (Table 3). Although there are less data with these approaches, they can also be considered. We would generally consider these regimens if he does not respond to reinduction or NECTAR. The phosphatidylinositol 3-kinase/AKT/mTOR pathway is frequently activated in T-ALL and preclinical data show efficacy of inhibition.80 The mTOR inhibitor everolimus, combined with 4-drug reinduction, showed promising activity in children with relapsed ALL; however, the vast majority had B-ALL relapses.81 The Cyclin D-CDK4/6 axis is implicated in T-cell leukemogenesis,82,83 and clinical trials are investigating the CDK4/6 inhibitor palbociclib combined with chemotherapy (NCT03792256, NCT03515200) and ribociclib with everolimus (NCT03740334). BCL2 inhibition is another strategy under investigation in relapsed T-ALL; venetoclax (selective BCL2 inhibitor) and navitoclax (BCL-2, BCL-XL and BCL-W inhibitor) have shown single-agent activity in preclinical models as well as synergistic activity in ALL xenografts.84-86 Early results from phase 1 trials with both venetoclax (NCT03236857)87 and combined venetoclax/navitoclax (NCT03181126)88 are intriguing. The overall response rate for venetoclax/navitoclax plus chemotherapy in children with r/r T-ALL/lymphoblastic lymphoma was 86% (6/7) with best responses of CR/CR with incomplete marrow recovery/CR with incomplete platelet recovery in 5 patients (71%).88
Conclusions
Until recently, the main treatment options for relapsed ALL were cytotoxic chemotherapy and HSCT. Now, there are a variety of treatment options, including highly active immunotherapies for B-ALL, small molecule inhibitors of pathways altered in relapsed B- and T-ALL, and improved HSCT technologies. Risk stratification has been further refined at initial diagnosis, and some of these therapies are now also being investigated in the frontline setting. We anticipate that, although this may influence outcomes at the time of relapse,89 survival will continue to improve for children and adolescents with relapsed and refractory ALL as a growing number of molecularly and immunologically targeted therapies are developed.
Authorship
Contribution: S.P.H. and E.A.R. wrote the paper.
Conflict-of-interest disclosure: S.P.H. has received consulting fees from Novartis, honoraria from Amgen, owns stock in Amgen, and is the Jeffrey E. Perelman Distinguished Chair in Pediatrics at the Children’s Hospital of Philadelphia. E.A.R. receives research funding (institutional) from Pfizer, serves on a DSMB for Celgene/BMS, and is a KiDS of NYU Foundation Professor of Pediatrics at NYU Grossman School of Medicine.
Correspondence: Stephen P. Hunger, CTRB Room 3060, 3501 Civic Center Blvd, Children’s Hospital of Philadelphia, Philadelphia, PA 19104; e-mail: hungers@email.chop.edu.