Key Points
Maternal hepcidin, but not embryo hepcidin, controls embryo iron endowment in mouse models under physiological conditions.
Hepcidin-mediated maternal iron restriction during pregnancy causes embryo anemia, tissue iron deficiency, and decreased weight.

Abstract
Iron disorders are associated with adverse pregnancy outcomes, yet iron homeostatic mechanisms during pregnancy are poorly understood. In humans and rodents, the iron-regulatory hormone hepcidin is profoundly decreased in pregnant mothers, which is thought to ensure adequate iron availability for transfer across placenta. However, the fetal liver also produces hepcidin, which may regulate fetal iron endowment by controlling placental iron export. To determine the relative contribution of maternal vs embryo hepcidin to the control of embryo iron endowment in iron-sufficient or iron-overloaded mice, we generated combinations of mothers and embryos that had or lacked hepcidin. We found that maternal, but not embryonic, hepcidin determined embryo and placental iron endowment in a healthy pregnancy. We further determined that inflammation can counteract pregnancy-dependent suppression of maternal hepcidin. To establish how essential maternal hepcidin suppression is for embryo iron homeostasis, we mimicked the range of maternal hepcidin activity by administering a hepcidin peptide mimetic to pregnant mice. This also allowed us to determine the effect of isolated maternal hepcidin excess on pregnancy, in the absence of other confounding effects of inflammation. Higher doses of hepcidin agonist caused maternal iron restriction and anemia, lower placenta and embryo weight, embryo anemia, and increased embryo mortality. Low agonist doses did not cause maternal anemia but still adversely affected the embryo, causing anemia, tissue iron deficiency (including in the brain), and decreased weight. Our studies demonstrate that suppression of maternal hepcidin during pregnancy is essential for maternal and embryo iron homeostasis and health.
Introduction
Systemic iron homeostasis is regulated by the hormone hepcidin,1 which triggers degradation of the only known mammalian iron exporter ferroportin (FPN). Decreased FPN levels prevent iron absorption, recycling, and mobilization from storage, thereby decreasing plasma iron levels and iron utilization in tissues.2 Major stimuli that regulate hepcidin production include iron, erythropoietic activity, pregnancy, and inflammation.3,4 Hepcidin suppression during iron deficiency allows for replenishment of body iron stores, and suppression after an erythropoietic stimulus allows sufficient delivery of iron for compensatory erythropoiesis. Inflammation increases hepcidin production via the interleukin-6 pathway,5,6 resulting in hypoferremia. Although this host defense mechanism may slow the growth of invading microorganisms, in chronic inflammatory conditions or infections, elevated hepcidin contributes to the development of iron-restricted anemia.7
During pregnancy, iron is critical for placental growth, fetal development, and maternal health.8 In humans, iron deficiency and its more severe form, iron deficiency anemia, have been associated with adverse pregnancy outcomes, including increased maternal morbidity and mortality, preterm birth, low birth weight, cognitive defects in newborns, and impaired immune function.9-14 In healthy human15 and rodent16,17 pregnancies, maternal hepcidin levels are profoundly decreased in the second and third trimester by an unknown mechanism but are still regulated by maternal iron status.16,18 Decreased maternal hepcidin allows for increased absorption of dietary iron and mobilization of iron from stores,19 thus increasing the availability of iron for transfer across the placenta.
Although maternal hepcidin is low in normal pregnancies, inflammatory disorders could prevent appropriate suppression of hepcidin, potentially compromising iron availability during pregnancy. In addition to causing iron sequestration in the mother, elevated maternal hepcidin could prevent iron absorption from supplements commonly prescribed to pregnant women. A study utilizing stable iron isotope administration in healthy pregnancies reported that higher maternal hepcidin was associated with lower dietary iron absorption and lower iron transfer to the neonate.20
Inflammation during pregnancy is common in developing countries with endemic malaria, HIV, tuberculosis, or parasitic infestations, whereas in high-resource countries, inflammation occurs in bacterial and viral infections, obesity, diabetes, and, less commonly, autoimmune diseases or malignancies.21 It has not been systematically explored whether maternal hepcidin is increased in these conditions, particularly in pregnancies with moderate to severe inflammation, how maternal hepcidin is regulated in the context of coexisting anemia and whether maternal hepcidin has any effect on neonatal iron homeostasis. In a cross-sectional cohort including both healthy and complicated pregnancies (gestational diabetes mellitus, gestational hypertension, preeclampsia and liver dysfunction), maternal hepcidin correlated with C-reactive protein22 but in healthy human pregnancy maternal serum hepcidin did not correlate with inflammatory markers.15,23,24 Maternal hepcidin predicted anemia of inflammation in pregnant women with schistosomiasis.25 In the milder inflammation associated with obesity, some studies reported no difference in maternal hepcidin compared with lean pregnancies in early pregnancy or the third trimester,26-28 whereas other studies found elevated maternal serum hepcidin in obese pregnant women.29-32 No clear association with neonatal iron status or anemia was reported. Importantly, in human studies utilizing iron parameters measured in cord blood, evaluation of fetal/neonatal iron homeostasis is confounded by the effects of labor and delivery. Indeed, a time-course analysis of perinatal hepcidin expression in mice showed that embryo hepcidin expression is low, but transiently increases at birth until at least postnatal day 2 (P2).33
In this study, we examined the role and regulation of hepcidin during pregnancy and found that suppression of maternal hepcidin during pregnancy is essential for embryo iron homeostasis and health.
Methods
Mice
All experiments were approved by the Animal Research Committee at University of California, Los Angeles (UCLA). Mice were fed a standard diet (PicoLab Rodent Diet 20, 5053 Irradiated, 185 ppm iron). All mouse strains were on a C57BL/6J background. Wild-type (WT) mice were from The Jackson Laboratory or bred in-house. Hepcidin knockout (KO) mice were originally from Sophie Vaulont19 and backcrossed by us onto the C57BL/6J background.
To induce maternal inflammation, pregnant WT mice received a subcutaneous interscapular injection of 0.5 μg/g lipopolysaccharide (LPS; Escherichia coli serotype O55:B5, MilliporeSigma) or sterile water at E17.5. To model iron restriction, pregnant C57BL/6J mice were injected subcutaneously with 10 or 50 nmol of minihepcidin (PR73) dissolved in SL22034,35 (NOF Corporation); controls were injected with the corresponding amount of solvent. Injections were given daily from E12.5 to E17.5 (6 days) or E7.5 to E17.5 (11 days). PR73 (molecular weight, 1730 Da) was synthesized as described previously.35
Statistical analysis
Statistical analysis was performed using GraphPad Prism 8 or SigmaPlot (Systat Software). Details are provided in the supplemental Methods (available on the Blood Web site).
Additional methods can be found in the supplemental Methods.
Results
Embryo iron endowment is determined by maternal rather than embryo or placental hepcidin
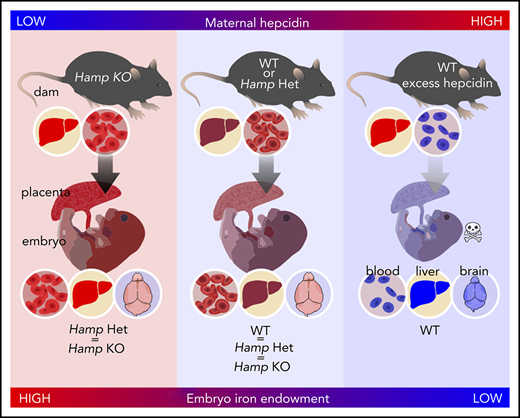
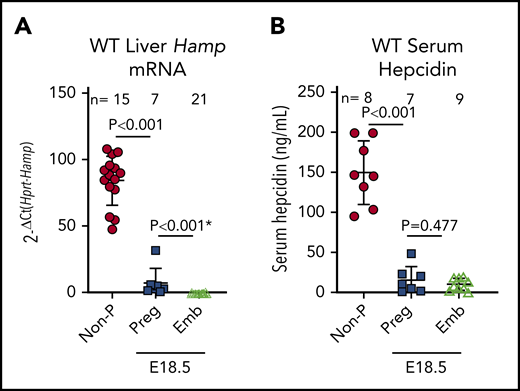
Maternal hepcidin (Hamp) is profoundly suppressed during human15 and rodent pregnancies,16,17 reaching maximal suppression in the third trimester when iron transfer to the fetus is greatest (reviewed in Koenig et al36 ). However, the fetal liver also produces hepcidin, which could control placental iron export through FPN. At embryonic day 18.5 (E18.5), maternal liver Hamp messenger RNA (mRNA) was 12-fold lower than in nonpregnant females (2−ΔCt 84 ± 18 vs 7 ± 10; P < .001), and embryo liver Hamp was another 15-fold lower than maternal Hamp (2−ΔCt 7 ± 10 vs 0.46 ± 0.49; P < .001) (Figure 1A). Serum hepcidin was also suppressed in pregnant females and embryos compared with nonpregnant females (150 ± 37 vs 16 ± 16 vs 11 ± 7 ng/mL for nonpregnant, pregnant females, and embryos, respectively (Figure 1B). Recently, we showed that during iron deficiency, embryo iron endowment was not affected by the embryo’s hepcidin genotype.16 However, under high-iron conditions, when hepcidin is typically induced,4 embryo hepcidin could limit iron transfer across the placenta. We examined the contribution of maternal and embryo hepcidin to regulating iron transfer from mother to placenta and embryo in normal and iron-loaded pregnancies. The iron-loaded Hamp KO and iron-sufficient Hamp heterozygous (Het) females were mated with Hamp Het males to generate genetically mixed litters of Hamp KO, Het, and WT embryos. Hamp KO females were significantly more iron-loaded than Het females at E18.5 (serum iron, 45 ± 2 vs 20 ± 8 ng/mL [P = .036; Figure 2A] and liver nonheme iron, 417 ± 131 vs 13 ± 6 µg/g P = .003; Figure 2B]). We then compared the iron status of embryos and their placentas at E18.5 within each of the maternal genotypes. Although embryo Hamp mRNA was generally low in all genotype groups at E18.5, statistically significant differences were detected in liver Hamp expression among WT, Het, and KO embryos (supplemental Figure 1A). These variations in expression were not functionally relevant, as there was no difference in embryo serum iron, embryo liver nonheme iron, and placental nonheme iron concentrations (P = .95, P = .75, and P = .88) among embryo Hamp genotypes derived from Het mothers or among embryos derived from KO mothers (P = .08, P = .29, and P = .17) (Figure 2C-E). These data demonstrate that even under high-iron conditions, embryo hepcidin has no effect on embryo iron endowment.

Hepcidin expression in nonpregnant and pregnant females and embryos in mice. WT C57BL6/J nonpregnant females were compared with age-matched pregnant females and their embryos at E18.5 for hepatic Hamp mRNA (A) and serum hepcidin (B) concentrations. Statistical differences between groups was determined by 2-tailed Student t test for normally distributed values or by Mann-Whitney rank-sum test for nonnormally distributed values (indicated by * following P value). Emb, embryos; Non-P, nonpregnant; Preg, pregnant.
Hepcidin expression in nonpregnant and pregnant females and embryos in mice. WT C57BL6/J nonpregnant females were compared with age-matched pregnant females and their embryos at E18.5 for hepatic Hamp mRNA (A) and serum hepcidin (B) concentrations. Statistical differences between groups was determined by 2-tailed Student t test for normally distributed values or by Mann-Whitney rank-sum test for nonnormally distributed values (indicated by * following P value). Emb, embryos; Non-P, nonpregnant; Preg, pregnant.
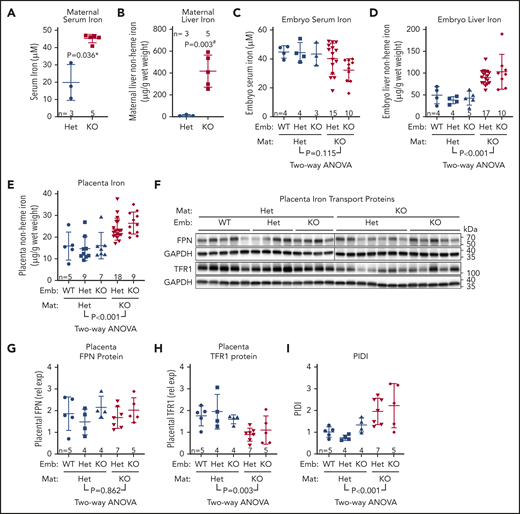
Maternal rather than embryo hepcidin determines embryo iron endowment. To determine the relative contribution of maternal vs embryo hepcidin to the regulation of embryo iron endowment, Hamp Het and KO females were mated with Hamp Het males to generate Hamp WT, Het, and KO embryos. Mothers and embryos were analyzed on E18.5 for maternal serum iron (A) and liver non-heme iron (B) concentrations. Statistical comparisons were performed by 2-tailed Mann-Whitney rank-sum test for nonnormally distributed values (indicated by * following P values) or 2-tailed t test with Welch’s correction for normally distributed but nonequal variance data sets (indicated by # after the P value) for embryo serum iron (C), embryo liver nonheme iron (D), and placenta nonheme iron (E) concentrations. (F) Western blot for FPN and TFR1 from E18.5 Hamp WT, Het, and KO placentas, derived from either Hamp Het mothers and Hamp KO mothers. GAPDH was used as a loading control. FPN was run on 2 separate blots, as indicated by a line; TFR1 was run on a single blot. (G) Quantification of placental FPN across different maternal and embryo genotypes. FPN was normalized to GAPDH for loading. To normalized FPN levels between blots, 4 samples from each of the 2 blots were rerun together on a separate blot. (H) Quantification of placental TFR1 across different maternal and embryo genotypes. TFR1 was normalized to GAPDH. (I) Calculated PIDI, the ratio of placental FPN to TFR1 protein. (C-E, G-I) Statistical differences between groups was determined by 2-way analysis of variance for embryo vs mother Hamp genotype. Mat, maternal.
Maternal rather than embryo hepcidin determines embryo iron endowment. To determine the relative contribution of maternal vs embryo hepcidin to the regulation of embryo iron endowment, Hamp Het and KO females were mated with Hamp Het males to generate Hamp WT, Het, and KO embryos. Mothers and embryos were analyzed on E18.5 for maternal serum iron (A) and liver non-heme iron (B) concentrations. Statistical comparisons were performed by 2-tailed Mann-Whitney rank-sum test for nonnormally distributed values (indicated by * following P values) or 2-tailed t test with Welch’s correction for normally distributed but nonequal variance data sets (indicated by # after the P value) for embryo serum iron (C), embryo liver nonheme iron (D), and placenta nonheme iron (E) concentrations. (F) Western blot for FPN and TFR1 from E18.5 Hamp WT, Het, and KO placentas, derived from either Hamp Het mothers and Hamp KO mothers. GAPDH was used as a loading control. FPN was run on 2 separate blots, as indicated by a line; TFR1 was run on a single blot. (G) Quantification of placental FPN across different maternal and embryo genotypes. FPN was normalized to GAPDH for loading. To normalized FPN levels between blots, 4 samples from each of the 2 blots were rerun together on a separate blot. (H) Quantification of placental TFR1 across different maternal and embryo genotypes. TFR1 was normalized to GAPDH. (I) Calculated PIDI, the ratio of placental FPN to TFR1 protein. (C-E, G-I) Statistical differences between groups was determined by 2-way analysis of variance for embryo vs mother Hamp genotype. Mat, maternal.
By contrast, there was a highly significant effect of maternal Hamp genotype on embryo iron endowment. Maternal hepcidin deficiency significantly increased concentration of embryo liver nonheme iron (P < .001 for maternal vs P = .377 for embryo Hamp genotype) and increased placenta nonheme iron (P < .001 for maternal vs P = .391 for embryo Hamp genotype) (Figure 2D-E). Similar to results at E18.5, analysis of neonatal liver nonheme iron at P1 demonstrated a highly significant effect from maternal, but not pup, genotype (P < .001 for maternal vs P = .915 for pup Hamp genotype) (supplemental Figure 1B). Our data in the mouse model demonstrate that the degree of maternal hepcidin suppression rather than embryo or placental hepcidin determines embryo iron endowment.
Placental FPN levels are a possible target of placental and embryo hepcidin, but we found no difference in placental FPN in relation to either embryo genotype (P = .219) or maternal Hamp genotype (P = .862) (Figure 2F-G). In contrast, TFR1 was decreased in placentas from Hamp KO dams (P = .003), with no difference observed between embryo Hamp genotypes (P = .993) (Figure 2F,H). The decrease in placental TFR1 is likely mediated by the iron responsive element (IRE)-iron regulatory protein (IRP) system in response to increased placenta iron concentration.
We previously conceived the placental iron deficiency index (PIDI), the ratio of placental FPN to TFR1 protein concentrations, as a sensitive indicator of placental iron status.16 In that study, we reported that PIDI was decreased in placentas from iron-deficient compared with iron-adequate mothers. We now show that PIDI increased with placental iron loading and was higher in placentas from Hamp KO compared with Het dams (P < .001), with no effect of embryo Hamp genotype (P = .337) (PIDI in Figure 2I, placental iron in Figure 2E).
There was no difference in embryo brain iron regardless of embryo or maternal genotype (P = .878 and P = .852) (supplemental Figure 1C).
Maternal inflammation induces maternal hepcidin
To assess whether inflammation could counteract the physiological suppression of maternal hepcidin during pregnancy, C57BL/6J females at E17.5 and nonpregnant controls were injected subcutaneously with 0.5 µg/g LPS for 6 hours. LPS treatment of dams is known to rapidly increase the concentrations of the cytokine interleukin-6,37 a potent inducer of hepcidin. In nonpregnant females, LPS induced hepcidin 3.5-fold compared with water-injected controls (198 ± 136 to 706 ± 352 ng/mL, P = .029, Figure 3A). In E17.5 pregnant females, serum hepcidin was lower at baseline than in nonpregnant females as expected but was induced 23-fold after LPS injection (37 ± 25 to 849 ± 279 ng/mL, P = .008), reaching levels observed in LPS-injected nonpregnant females (P = .496 for pregnant vs nonpregnant LPS-injected groups) (Figure 3A).
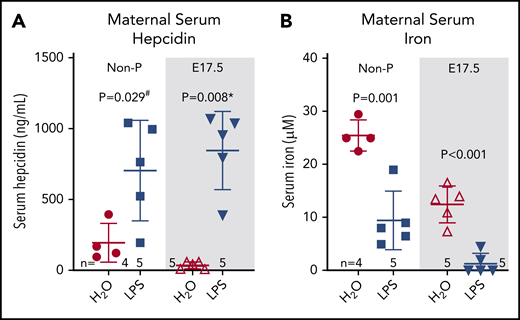
Systemic inflammation induces maternal hepcidin and severe hypoferremia during pregnancy. Age-matched nonpregnant and pregnant (E17.5) WT C57BL/6J females received a single subcutaneous injection of water or 0.5 µg/g LPS and were analyzed 6 hours later for maternal serum hepcidin (A) and maternal serum iron (B). Statistical comparisons were performed by 2-tailed Student t test for normally distributed values, 2-tailed Mann-Whitney rank-sum test for nonnormally distributed value (indicated by * following P-values), or 2-tailed t test with Welch’s correction for normally distributed but nonequal variance data sets (indicated by # after the P value).
Systemic inflammation induces maternal hepcidin and severe hypoferremia during pregnancy. Age-matched nonpregnant and pregnant (E17.5) WT C57BL/6J females received a single subcutaneous injection of water or 0.5 µg/g LPS and were analyzed 6 hours later for maternal serum hepcidin (A) and maternal serum iron (B). Statistical comparisons were performed by 2-tailed Student t test for normally distributed values, 2-tailed Mann-Whitney rank-sum test for nonnormally distributed value (indicated by * following P-values), or 2-tailed t test with Welch’s correction for normally distributed but nonequal variance data sets (indicated by # after the P value).
Following the hepcidin increase, we observed a 2.5-fold decrease in serum iron in nonpregnant LPS-treated females (25.5 ± 2.9 to 9.5 ± 5.5 µM, P = .001) (Figure 3B). In pregnant females, baseline serum iron was lower compared with nonpregnant females and was decreased further 9.5-fold from 12.5 ± 3.4 to 1.3 ± 2.0 µM following LPS treatment (P < .001, Figure 3B). Inflammatory hypoferremia is predominantly mediated by elevated hepcidin concentrations,38 although direct suppression of Fpn mRNA expression by inflammation39 may also contribute. We next explored the effect of such severe hypoferremia on maternal, placental, and fetal health, apart from other effects of inflammation.
Elevated maternal hepcidin results in maternal iron restriction
Maternal infection and inflammation are associated with adverse pregnancy outcomes,40,41 but whether hepcidin-mediated hypoferremia contributes to those outcomes is unknown. To isolate the effect of elevated maternal hepcidin on pregnancy from other effects of inflammation, we injected pregnant mice with a hepcidin peptide mimetic PR73.34,42 To first determine whether PR73 crosses the placenta, mothers received a single subcutaneous injection of 100 nmol PR73 or solvent, and maternal and embryo sera were collected after 6 hours for an in vitro bioassay.43 HEK293T-FPN-GFP cells were treated with 10% of maternal or embryo serum in Dulbecco’s modified Eagle medium, and FPN levels determined by flow cytometry and compared with PR73 and hepcidin-25 standard curves (supplemental Figure 2A-B). Maternal serum from PR73-injected mothers caused degradation of FPN, but embryo serum from the same mothers did not, indicating that PR73 does not appreciably cross the placenta.
To examine pregnancy outcomes after a prolonged increase in hepcidin activity, WT pregnant females received daily subcutaneous injections of minihepcidin PR73 as follows: a high short-term dose (“50-6d," 50 nmol/day for 6 days from E12.5 to E17.5), a high long-term dose (“50-11d," 50 nmol/day for 11 days from E7.5 to E17.5), and a low long-term dose (“10-11d," 10 nmol/day for 11 days from E7.5 to E17.5). Pregnant female controls were injected on the same schedule with solvent. Analyses were performed at E18.5.
To estimate how much endogenous hepcidin the PR73 treatment is equivalent to, we assessed FPN degradation with the in vitro bioassay using maternal serum from the 50-6d treatment group (supplemental Figure 2C) and compared it to the hepcidin-25 standard curve. We calculated the average activity in maternal serum using the equation in supplemental Figure 2A and accounting for the use of 10% serum. The activity in 50-6d maternal serum was functionally equivalent to ∼700 ng/mL of hepcidin-25, which is comparable to endogenous maternal hepcidin levels observed in LPS-treated females (Figure 3). We also assessed FPN degradation by western blotting after treating FPN-GFP cells with maternal and embryo serum from our “50-6d” and “10-11d” treatment groups (supplemental Figure 2D). We found that maternal serum caused much greater FPN degradation than embryo serum, further indicating that PR73 did not appreciably cross the placenta in these treatment groups. We did not have sufficient material to analyze maternal and embryo sera from the 50-11d treatment group and thus cannot rule out that minihepcidins entered the embryo circulation in this group.
We next assessed the effect of increased hepcidin activity during pregnancy on maternal iron and hematological parameters. Minihepcidin treatment tended to decrease maternal serum iron concentrations in all groups, although because of measurement variability and limited sample sizes, statistical significance was only reached in the 50-6d group (Figure 4A). PR73 did cause a dose- and time-dependent iron sequestration in the maternal liver and spleen (Figure 4B-C), with the strongest effect seen in the 50-11d group. The same 50-11d group had the most severe anemia, with significantly decreased hemoglobin, red blood cell (RBC) count, and mean corpuscular volume (MCV) (Figure 4D-F). The effect on maternal iron and hematology was less pronounced in the 10-11d group that received the low dose of PR73, where dams did not develop anemia.
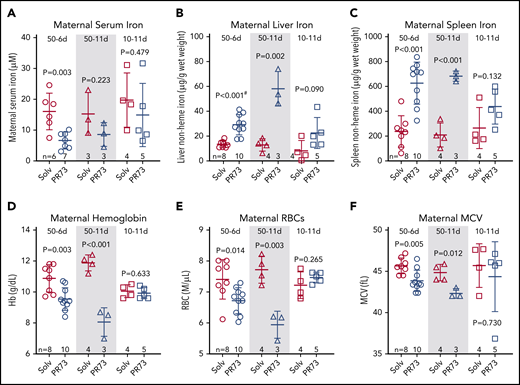
Effects of hepcidin-mediated iron restriction during pregnancy on maternal iron status and hematological parameters. Pregnant C57BL/6J dams received daily subcutaneous PR73 injections of 50 nmol/day for 6 days from E12.5 to E18.5 (50-6d), 50 nmol/day for 11 days from E7.5 to E17.5 (50-11d), 10 nmol/day for 11 days from E7.5 to E17.5 (10-11d) or the equivalent amount of solvent. Iron status and hematological parameters were measured at E18.5. Maternal iron parameters included serum iron (A), liver nonheme iron (B), and spleen nonheme iron (C). Maternal hematological parameters included hemoglobin (Hb) (D), RBC count (E), and MCV (F). Statistical comparisons were performed by 2-tailed Student t test for normally distributed values or for normally distributed but nonequal variance data sets, a 2-tailed t test with Welch’s correction was performed (indicated by # after the P value).
Effects of hepcidin-mediated iron restriction during pregnancy on maternal iron status and hematological parameters. Pregnant C57BL/6J dams received daily subcutaneous PR73 injections of 50 nmol/day for 6 days from E12.5 to E18.5 (50-6d), 50 nmol/day for 11 days from E7.5 to E17.5 (50-11d), 10 nmol/day for 11 days from E7.5 to E17.5 (10-11d) or the equivalent amount of solvent. Iron status and hematological parameters were measured at E18.5. Maternal iron parameters included serum iron (A), liver nonheme iron (B), and spleen nonheme iron (C). Maternal hematological parameters included hemoglobin (Hb) (D), RBC count (E), and MCV (F). Statistical comparisons were performed by 2-tailed Student t test for normally distributed values or for normally distributed but nonequal variance data sets, a 2-tailed t test with Welch’s correction was performed (indicated by # after the P value).
To evaluate the maternal response to iron restriction and anemia in the 50 nmol/day injection groups, we first confirmed that PR73 treatment did not induce inflammation, as reflected by the stable liver serum amyloid A1 (Saa1) mRNA (supplemental Figure 3A). Endogenous maternal liver Hamp mRNA was further suppressed, particularly in the 50-11d group (P = .044) (supplemental Figure 3B), likely as a feedback response to iron restriction. To evaluate the erythropoietic response, we measured mRNA expression of maternal bone marrow glycophorin A (Gypa) and erythroferrone (Erfe) (supplemental Figure 3C-D). Erfe, but not Gypa, was significantly elevated in 50-6d and 50-11d groups, suggesting an ineffectual erythropoietic response to anemia. Our data show that despite adequate dietary iron, elevated maternal hepcidin activity during pregnancy caused maternal iron restriction and maternal anemia.
Effects of hepcidin-mediated maternal iron restriction on the placenta
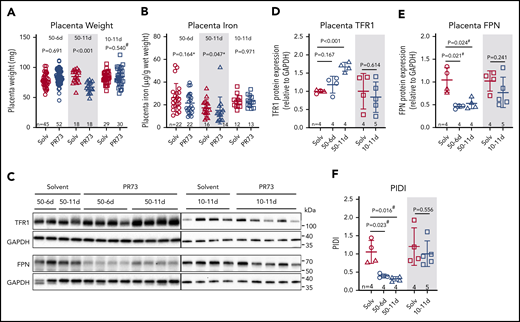
Placentas were analyzed at E18.5 to determine the consequences of maternal iron restriction on placenta development and iron status. In the 50-6d treatment group, injections started at E12.5, after placental development is complete.44 There was no difference in placenta weight (P = .691, Figure 5A) or nonheme iron concentration (P = .164, Figure 5B) between minihepcidin-treated and control pregnancies. In the 50-11d treatment group, where injections started at E7.5, before complete placental development, placental weights at E18.5 were 20% lower in the minihepcidin-treated group (P < .001, Figure 5A). Decreased placental weight suggests impaired placental development, which may impair nutrient transport to the embryo, not limited to iron. Additionally, placental nonheme iron concentrations were significantly lower in this group (P = .047, Figure 5B), despite adaptive alterations in expression of iron transporters TFR1 and FPN (see below). In the 10-11d treatment group, despite injections starting prior to placental maturation, there was no difference in placental weight or placental nonheme iron concentration (Figure 5A-B), indicating that the level of iron restriction was insufficient to affect placental development.
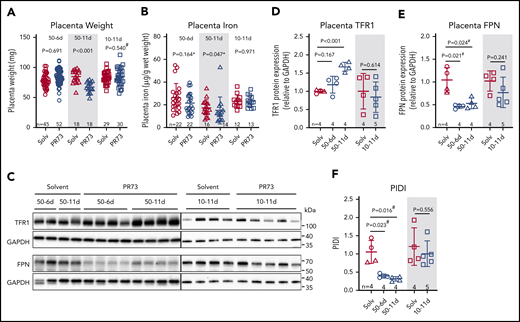
Effects of hepcidin-mediated maternal iron restriction on the placenta. Placentas from the 3 minihepcidin treatment groups from Figure 4 were analyzed at E18.5 for weight and iron content. (A) Placental weight was used as a surrogate measure for placental development. (B) Placental nonheme iron content. (C) Western blot of total placental TFR1 and FPN protein for the 50-6d and 50-11d treatment groups (left panel) and 10-11d treatment group (right panel). Quantification of western blot results for TFR1 (D) and FPN (E) protein relative to GAPDH. (F) Calculated PIDI, the ratio of placental FPN to TFR1 protein. Statistical comparisons were performed by 2-tailed Student t test for normally distributed values, 2-tailed Mann-Whitney rank-sum test for nonnormally distributed values (indicated by * following P-values), or 2-tailed t test with Welch’s correction for normally distributed but nonequal variance data sets (indicated by # after the P value).
Effects of hepcidin-mediated maternal iron restriction on the placenta. Placentas from the 3 minihepcidin treatment groups from Figure 4 were analyzed at E18.5 for weight and iron content. (A) Placental weight was used as a surrogate measure for placental development. (B) Placental nonheme iron content. (C) Western blot of total placental TFR1 and FPN protein for the 50-6d and 50-11d treatment groups (left panel) and 10-11d treatment group (right panel). Quantification of western blot results for TFR1 (D) and FPN (E) protein relative to GAPDH. (F) Calculated PIDI, the ratio of placental FPN to TFR1 protein. Statistical comparisons were performed by 2-tailed Student t test for normally distributed values, 2-tailed Mann-Whitney rank-sum test for nonnormally distributed values (indicated by * following P-values), or 2-tailed t test with Welch’s correction for normally distributed but nonequal variance data sets (indicated by # after the P value).
We previously reported that during maternal iron deficiency, placental iron transporters are regulated to maintain placental iron homeostasis.16 As placental tissue becomes iron deficient, placental IRP-1 mediates an increase in TFR1 and decrease in FPN protein, protecting placental tissues from iron deficiency.16 Maternal iron restriction resulted in similar regulation of placental iron transporters (Figure 5C-E). In the most severely iron-restricted group (50-11d), TFR1 significantly increased (P < .001) (Figure 5C-D) and FPN decreased (P = .024) (Figure 5C,E). PIDI (the FPN/TFR1 ratio) was significantly decreased in the 50-6d and 50-11d PR73 groups, indicating the placentas “sensed” iron restriction and responded by altering the transporter expression (Figure 5F) to preserve their iron homeostasis. These transporter changes, however, were not sufficient to prevent placental nonheme iron decrease in the face of severe maternal iron restriction. In the shorter term 50-6d group, only FPN significantly decreased (P = .021), which was sufficient to maintain normal placental nonheme iron levels. In the 10-11d group, neither TFR1 nor FPN nor PIDI differed significantly between the solvent and minihepcidin groups, consistent with the mild iron restriction (Figure 5D-F).
Hepcidin-mediated maternal iron restriction causes adverse embryo outcomes
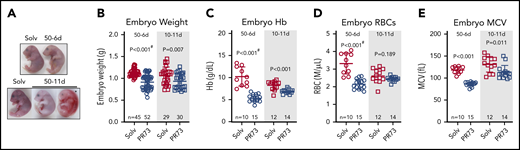
Embryos from minihepcidin- and solvent-treated mothers were analyzed at E18.5. In the most severe 50-11d group, in which placental development was also perturbed, 40% of embryos were dead and resorbing, and surviving embryos exhibited marked pallor (Figure 6A). Such severe adverse outcomes highlight the detrimental effects of maternal iron restriction on pregnancy and may result not only from iron deficiency in the embryo itself but also the indirect effects of iron restriction via maternal anemia or placental insufficiency. Because of frequent embryo death in this group, further analyses were performed in the embryos from the milder 50-6d and 10-11d minihepcidin groups. Embryos weighed significantly less in both the 50-6d and 10-11d groups compared with the solvent groups (50-6d: 1.13 ± 0.08 vs 0.92 ± 0.16 g, P < .001; 10-11d: 1.07 ± 0.19 vs 0.93 ± .017 g, P = .007) (Figure 6B), indicating that embryos are much more susceptible to the detrimental effect of iron restriction than the mothers or placentas. Furthermore, both treatments caused embryo anemia. A stronger effect was noted in the 50-6d group, where embryo hemoglobin, RBC number, and MCV were significantly lower compared with solvent group (all P < .001, Figure 6C-E). In the 10-11d groups, embryo Hb and MCV were significantly lower (P < .001 and P = .011, Figure 6C,E); but RBC counts did not statistically differ (P = .189, Figure 6D).
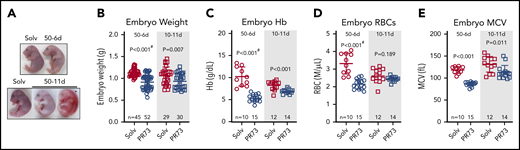
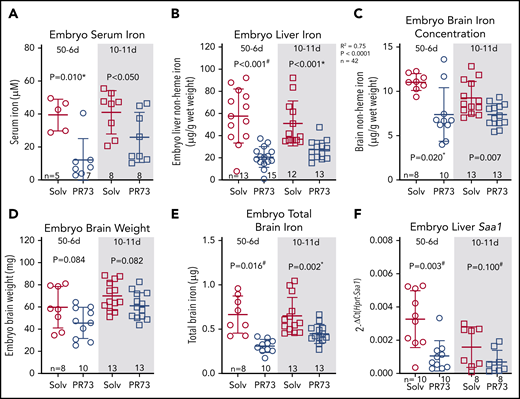
Effects of hepcidin-mediated maternal iron restriction on embryo outcome and hematological parameters. Embryos from PR73- and solvent-injected pregnancies from Figure 4 were analyzed at E18.5. (A) Embryos were visibly anemic, with increased mortality and resorption in the more severe 50-11d group. (B-E) Analysis of embryos from the 50-6d and 10-11d groups. (B) Embryo weight. (C-E) Blood hemoglobin concentration (C), RBC count (D), and mean corpuscular volume MCV (E). Statistical comparisons were performed by 2-tailed Student t test for normally distributed values or 2-tailed t test with Welch’s correction for normally distributed but non-equal variance data sets (indicated by # after the P value).
Effects of hepcidin-mediated maternal iron restriction on embryo outcome and hematological parameters. Embryos from PR73- and solvent-injected pregnancies from Figure 4 were analyzed at E18.5. (A) Embryos were visibly anemic, with increased mortality and resorption in the more severe 50-11d group. (B-E) Analysis of embryos from the 50-6d and 10-11d groups. (B) Embryo weight. (C-E) Blood hemoglobin concentration (C), RBC count (D), and mean corpuscular volume MCV (E). Statistical comparisons were performed by 2-tailed Student t test for normally distributed values or 2-tailed t test with Welch’s correction for normally distributed but non-equal variance data sets (indicated by # after the P value).
We next assessed iron status of embryos. Embryos were hypoferremic in both groups; in the 50-6d group, serum iron decreased >3-fold (39.7 ± 8.6 vs 12.4 ± 12.0 µM, P = .010), and in the 10-11d group, serum iron decreased from 41.3 ± 12.3 to 26 ± 14.2 µM (P < .05) (Figure 7A). Additionally, iron stores assessed by embryo liver nonheme iron concentration were significantly decreased (50-6d, 58 ± 23 to 21 ± 9 µg/g [P < .001]; 10-11d, 51 ± 19 to 27 ± 8 µg/g [P < .001]) (Figure 7B). Brain nonheme iron concentration was also significantly decreased following maternal iron restriction (Figure 7C). The slight trend for lower brain weight in the iron-restricted groups was not statistically significant (Figure 7D). Taking into account embryo brain weight, total brain iron was also significantly decreased in embryos from iron-restricted mothers (Figure 7E). There was no evidence of embryo inflammation in the minihepcidin-treated group as measured by embryo liver Saa1 mRNA (Figure 7F). Supplemental Figure 4 provides a graphical summary of the consequences of elevated maternal hepcidin. Our data in the mouse model demonstrate that even modest hepcidin-mediated iron restriction, which did not substantially alter maternal iron and hematological status or placental homeostasis, still had a detrimental effect on embryos, manifested as growth restriction, anemia, and tissue iron deficiency, including in the brain.
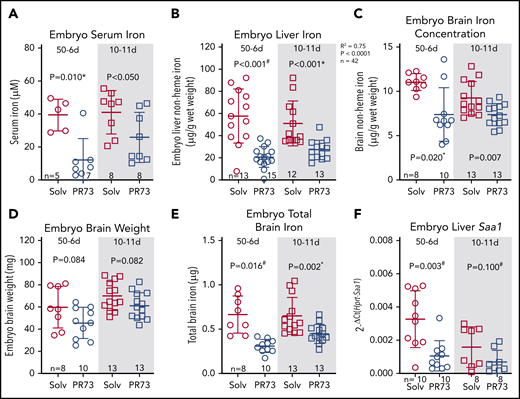
Effects of hepcidin-mediated maternal iron restriction on embryo iron status. Embryos from PR73- and solvent-injected pregnancies from Figure 4 were analyzed at E18.5. (A) Serum iron. (B) Embryo liver non-heme iron. (C) Embryo brain nonheme iron concentration. (D) Embryo brain weight. (E) Total embryo brain nonheme iron content. (F) Embryo liver Saa1 mRNA expression. Statistical comparisons were performed by 2-tailed Student t test for normally distributed values, 2-tailed Mann-Whitney rank-sum test for nonnormally distributed values (indicated by * following P-values), or 2-tailed t test with Welch’s correction for normally distributed but nonequal variance data sets (indicated by # after the P value).
Effects of hepcidin-mediated maternal iron restriction on embryo iron status. Embryos from PR73- and solvent-injected pregnancies from Figure 4 were analyzed at E18.5. (A) Serum iron. (B) Embryo liver non-heme iron. (C) Embryo brain nonheme iron concentration. (D) Embryo brain weight. (E) Total embryo brain nonheme iron content. (F) Embryo liver Saa1 mRNA expression. Statistical comparisons were performed by 2-tailed Student t test for normally distributed values, 2-tailed Mann-Whitney rank-sum test for nonnormally distributed values (indicated by * following P-values), or 2-tailed t test with Welch’s correction for normally distributed but nonequal variance data sets (indicated by # after the P value).
Discussion
The iron supply to the developing fetus is essential for its development and is entirely dependent on placental transfer of iron from the maternal circulation. Plasma iron levels are controlled by the iron-regulatory hormone hepcidin. Maternal hepcidin is profoundly decreased starting in the second trimester and until delivery in humans45 and rodents,16,17 presumably to mobilize maternal iron for transfer to the fetus. The relative contribution of maternal, fetal, and placental hepcidin to iron regulation in the mother and in the placentofetal unit during physiologic pregnancy is still not completely understood. In embryos with transgenic hepcidin overexpression under the control of the transthyretin promoter that is active as early as E9.5, severe embryo anemia was observed, with perinatal mortality.33 Similarly, in Tmprss6−/− embryos, hepcidin was elevated by E17.5 and was associated with iron deficiency and anemia in embryos and pups,46 but not embryonic lethality.47 Of note, dams in Tmprss6 studies were Het for Tmprss6 deletion, as homozygosity causes infertility in females.47 Conversely, in mice with targeted disruption of the hepcidin gene, both genders were fertile, and hepcidin KO pups were born at the expected Mendelian ratios and appeared normal.19 These genetic mouse models established that elevated embryo hepcidin is detrimental, whereas absence of embryo hepcidin does not cause obvious developmental defects. However, the regulatory role of embryonic hepcidin in physiologic pregnancy had not been determined.
During maternal iron loading, we assessed whether a physiological induction of embryonic hepcidin could be functionally important to prevent embryo iron overload in utero. We found no effect of embryo or placental hepcidin on embryo iron endowment in either iron-sufficient or iron-overloaded dams. Taken together with our previous finding that neither embryo nor placental hepcidin plays a role during maternal iron deficiency,16 we surmise that maternal hepcidin is a key determinant of embryo iron endowment. This is further evidenced by increased embryo iron endowment, regardless of embryo hepcidin genotype, when maternal hepcidin was absent and mothers were iron loaded. The importance of maternal hepcidin in determining embryo iron endowment is also supported by our previous findings that in WT dams with high maternal iron stores, maternal hepcidin was proportionally increased, limiting the release of stored maternal iron and preventing embryo iron overload.16 It is important to note however that in pathological pregnancies, embryo hepcidin can be increased, resulting in dysregulated iron homeostasis.46,48 In mice, maternal systemic inflammation induced by LPS injection also caused fetal hepcidin increase and hypoferremia within 24 hours.48 In rhesus macaques, intraamniotic injection of LPS similarly induced fetal hepcidin and hypoferremia, without any effect on maternal hepcidin.48 In human pregnancies, antenatal exposure to intraamniotic infections resulted in elevated cord blood hepcidin and accompanying hypoferremia.48 The strong regulation of fetal hepcidin by inflammation may have evolved as a conserved host defense mechanism against microbial infection of the fetoplacental unit. Taken together, data from our mouse models demonstrate that in healthy pregnancies or those with maternal iron loading, maternal hepcidin is the primary physiological controller of embryo iron endowment, but that in certain genetic conditions (eg, Tmprss6 mutations) or with exposure to inflammation, fetal hepcidin can contribute to fetal iron regulation.
In the current study, inflammation in pregnant dams robustly induced maternal hepcidin despite signals that suppress maternal hepcidin during normal pregnancy. In humans, inflammatory markers during pregnancy correlated with maternal hepcidin expression in some studies that included complicated pregnancies,22,36,49 but the association was attenuated in pregnancies with mild inflammation, such as that caused by obesity.26-32 Another study documented an ∼15-fold increase of hepcidin in a subgroup with anemia of chronic inflammation compared with normal pregnancy.50 However, no effect of elevated maternal hepcidin on neonatal outcomes has been conclusively demonstrated. Increased maternal hepcidin was reported in first-trimester spontaneous abortion, although it is unclear if the increase is a cause or consequence of spontaneous abortion.18 Currently, multiple factors limit our understanding of the pathophysiology of hepcidin induction by inflammation in human pregnancy, including exclusion of pregnant women with severe infection or inflammation, inadequate definition of the normal range and interpretation of inflammatory biomarkers in pregnancy, and the complexity of hepcidin regulation in the setting of inflammation and pregnancy coexisting with iron deficiency and anemia. In our mouse model, LPS administration caused >20-fold induction of maternal hepcidin, which was accompanied by severe hypoferremia, raising the possibility that increased maternal hepcidin during inflammation could contribute to the known adverse effects of maternal inflammation on the fetus. In human pregnancy, inflammation has been linked to increased maternal and neonatal mortality and morbidity, with adverse outcomes including spontaneous abortion, preterm birth, intrauterine growth restriction, and neurodevelopmental impairment.51
To differentiate the effects of elevated maternal hepcidin from the other effects of inflammation on embryo development, pregnant dams were treated daily with injections of the hepcidin mimetic PR73. Other models of elevated hepcidin were not suitable as female mice deficient in matriptase-2 (Tmprss6), a negative regulator of hepcidin, are infertile.52 Elevated maternal hepcidin activity resulted in maternal iron restriction and detrimental effects on the pregnancy. The higher PR73 dose regimens (50-6d and 50-11d treatments) caused iron sequestration in maternal liver and spleen and hypochromic, microcytic anemia in dams. Unsurprisingly, embryos were also affected. Compared with the solvent groups, embryos were smaller, anemic, and iron deficient. The most severe iron restriction (50-11d) also resulted in decreased placental weight and death of 40% of embryos. At this dose, it is possible that placental dysfunction impaired general nutrient delivery to the embryo, contributing to the negative embryo outcomes. Likewise, maternal anemia and decreased oxygen delivery may have affected placental function. Remarkably, even at a low PR73 dose (10-11d treatment) that did not cause any appreciable change in maternal hematological parameters or placental development, we still observed adverse effects on embryos, with decreased embryo weight, anemia, and iron deficiency, highlighting the sensitivity of the embryo to maternal iron restriction. Our data suggest that elevated maternal hepcidin and consequent maternal iron restriction may be an unrecognized pathogenic factor in inflamed pregnancies that could contribute to fetal harm or demise.
Fetal brain development is highly influenced by the maternal environment. Maternal inflammation during pregnancy is associated with increased risk of neurodevelopmental disorders in offspring.53 Similarly, maternal iron deficiency is also associated with increased risk of neurobehavioral impairments.11,54 Whether iron restriction consequent of inflammation mimics iron deficiency, thus resulting in impaired brain development, has not been investigated. Interestingly, in our model of maternal iron restriction from excess maternal hepcidin, embryo brains were significantly smaller and contained less iron. The embryo brains were affected even when maternal iron and hematological parameters were unaffected, suggesting that the brain is particularly sensitive to decreases in iron supply.
We previously reported that during maternal iron deficiency, placental iron transporters are regulated to maintain placental iron homeostasis.16 In that report, we devised the PIDI, a biological measure of iron deficiency of the maternofetal unit. PIDI is the ratio of placenta FPN to TFR1, predominantly located on the fetal-facing and maternal-facing syncytiotrophoblast membranes, respectively. Lower values of PIDI indicate greater placental iron deficiency, and the index is expected to be less sensitive to changes in placental size, cellular composition, or sampling location than the measurement of a single iron transporter would be. In the 50-6d and 50-11d treatment groups, PIDI was lower in the PR73-treated compared with solvent groups, indicating placental sensing of iron restriction. Importantly, PIDI also changed with placental iron loading and was increased in placentas from Hamp KO compared with Hamp Het dams, consistent with the observed increase in placental iron content in Hamp KO dams. This demonstrated that PIDI changes across the entire spectrum of placental iron exposure, supporting its utility as a sensitive and biologically relevant indicator of maternofetal iron status.
Our study demonstrates that the maintenance of appropriate maternal hepcidin levels during pregnancy is essential for embryo iron homeostasis and health. In contrast to maternal hepcidin, embryo and placental hepcidin have no observable role in iron regulation in the absence of embryo inflammation. Future studies in pregnant women should reveal whether the combination of increased maternal serum hepcidin and hypoferremia is a useful marker of embryo iron restriction and its adverse consequences.
For original data, please e-mail the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Victoria Gabayan and Erika Valore for their assistance with methods development.
The work was supported by the National Institutes of Health, Eunice Kennedy Shriver National Institute of Child Health and Human Development (grant R01HD096863) (E.N.), the Executive Advisory Board of the Iris Cantor-UCLA Women’s Health Center, National Center for Advancing Translational Sciences UCLA Clinical and Translational Science Institute (grant UL1TR000124) (E.N.) and a pilot award (V.S.), and National Institutes of Health Ruth L. Kirschstein National Research Service Award (T32-5T32HL072752-13) (V.S.). Flow cytometry was performed in the UCLA Jonsson Comprehensive Cancer Center and Center for AIDS Research Flow Cytometry Core Facility, which is supported by National Institutes of Health awards (P30 CA016042 and 5P30 AI028697), and by the Jonsson Comprehensive Cancer Center, the UCLA AIDS Institute, the David Geffen School of Medicine at UCLA, the UCLA Chancellor’s Office, and the UCLA Vice Chancellor’s Office of Research.
Authorship
Contribution: V.S. designed and performed experiments, analyzed data, and wrote the manuscript; A.L.F. performed experiments and assisted with data interpretation; K.J.C. performed experiments; P.R. synthesized minihepcidins and assisted with interpretation of data; and T.G. and E.N. conceived the project, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: T.G. and E.N. are shareholders of and scientific advisors for Intrinsic LifeSciences and Silarus Therapeutics and are consultants for Ionis Pharmaceuticals, Protagonist Therapeutics, Akebia, Vifor Pharma, and Disc Medicine. The remaining authors declare no competing financial interests.
Correspondence: Elizabeta Nemeth, Center for Iron Disorders, Department of Medicine, David Geffen School of Medicine, University of California, Los Angeles, 10833 LeConte Ave, CHS 43-229, Los Angeles, CA 90095; e-mail: enemeth@mednet.ucla.edu.