Key Points
Exocyst interacts with BLOC-2 in the delivery of endosomal cargo to maturing Weibel Palade bodies essential for VWF multimerization.
Exocyst serves as a clamp, impeding VWF exocytosis, which can be reversibly inhibited to facilitate VWF release.

Abstract
von Willebrand factor (VWF) is an essential hemostatic protein that is synthesized in endothelial cells and stored in Weibel-Palade bodies (WPBs). Understanding the mechanisms underlying WPB biogenesis and exocytosis could enable therapeutic modulation of endogenous VWF, yet optimal targets for modulating VWF release have not been established. Because biogenesis of lysosomal related organelle-2 (BLOC-2) functions in the biogenesis of platelet dense granules and melanosomes, which like WPBs are lysosome-related organelles, we hypothesized that BLOC-2–dependent endolysosomal trafficking is essential for WPB biogenesis and sought to identify BLOC-2–interacting proteins. Depletion of BLOC-2 caused misdirection of cargo-carrying transport tubules from endosomes, resulting in immature WPBs that lack endosomal input. Immunoprecipitation of BLOC-2 identified the exocyst complex as a binding partner. Depletion of the exocyst complex phenocopied BLOC-2 depletion, resulting in immature WPBs. Furthermore, releasates of immature WPBs from either BLOC-2 or exocyst-depleted endothelial cells lacked high-molecular weight (HMW) forms of VWF, demonstrating the importance of BLOC-2/exocyst-mediated endosomal input during VWF maturation. However, BLOC-2 and exocyst showed very different effects on VWF release. Although BLOC-2 depletion impaired exocytosis, exocyst depletion augmented WPB exocytosis, indicating that it acts as a clamp. Exposure of endothelial cells to a small molecule inhibitor of exocyst, Endosidin2, reversibly augmented secretion of mature WPBs containing HMW forms of VWF. These studies show that, although BLOC-2 and exocyst cooperate in WPB formation, only exocyst serves to clamp WPB release. Exocyst function in VWF maturation and release are separable, a feature that can be exploited to enhance VWF release.
Introduction
von Willebrand Factor (VWF) is an essential plasma hemostatic factor synthesized and released from endothelial cells. von Willebrand disease, a deficiency state of mature plasma VWF, is the most common bleeding disorder in humans.1 Conversely, high VWF plasma levels or augmented VWF endothelial release is associated with increased cardiovascular morbidity.1-3 VWF also participates in other pathophysiologic processes such as angiogenesis and tumor metastasis.4-6 VWF is stored in specialized endothelial storage granules, Weibel Palade bodies (WPBs). Biogenesis of WPB and maturation of VWF are interdependent processes. Heterologous expression of VWF in nonendothelial cells results in generation of WPB-like organelles expressing components of trafficking machinery.7-9 Conversely, deficiency in known components of WPB trafficking machinery leads to altered VWF multimerization and exocytosis.10
The origin of WPBs is highly complex and incompletely understood. WPBs arise from the trans-Golgi network (TGN) by tubulation of the TGN limiting membrane induced by newly synthesized VWF oligomers, a process that requires clathrin and adaptor protein 1 (AP1).11-14 After formation, WPBs obtain cargoes from endosomes, although little is known about the mechanisms by which WPBs interact with endosomes. WPBs share several characteristics with lysosomal-related organelles (LROs), cell-specific specialized organelles that include platelet dense granules, melanosomes, and lytic granules of the cytotoxic-T cells, among others.15-17 Abnormal biogenesis of LROs is the underlying defect in Hermansky-Pudlak syndrome (HPS). HPS is a group of autosomal recessive disorders characterized by albinism, secondary to deficient melanin pigmentation, and bleeding, believed to be primarily due to lack of platelet dense granules.17 There are at least 10 confirmed human subtypes of HPS. The affected genes encode subunits of 4 protein complexes: AP3 and biogenesis of LRO complex (BLOC) 1, 2, and 3.18 The function of these cytoplasmic protein complexes in LRO biogenesis has primarily been extrapolated from melanosome biogenesis. Many individuals with HPS and murine models of HPS are known to have low plasma VWF antigen levels and decreased high-molecular-weight (HMW) multimers.19-21 Depletion of BLOC-2 (an obligate complex of HPS3, HPS5, and HPS6) in endothelial cells impairs secretagogue-induced exocytosis but its mechanism remains unknown.22 BLOC-2 targets endosomal tubular transport carriers to maturing melanosomes in melanocytes and has been deduced to function as a tether.23
Traffic between 2 membrane-bound compartments, such as endosome-derived cargo vesicles and maturing secretory granules, requires docking of cargo vesicles at target membranes before fusion, regulated by multisubunit tethering complexes.24 The octameric exocyst complex is one such tether, which is known to interact with SNAREs, Sec/Munc proteins, and small Rho and Rab GTPases, primarily in yeast.25,26 There is a growing literature placing exocyst in mammalian endosomes,27-32 but its function in endothelial cells, specifically WPB biogenesis and exocytosis, remains unexplored.
Here, we report that BLOC-2 is directly involved in transport of endosomal cargo to maturing WPBs. BLOC-2 depletion results in trapping of immature WPBs in a post-TGN compartment, impairing VWF multimerization. We further show that BLOC-2 interacts with the exocyst complex in its essential role in WPB biogenesis. Depletion of the exocyst complex, which can be found on endosomes, also disrupts WPB biogenesis and VWF multimerization. However, in contrast to BLOC-2 depletion, which impairs VWF exocytosis, exocyst depletion augments VWF exocytosis. A reversible small molecule inhibitor of exocyst, termed Endosidin2, augments release of VWF with a normal multimerization pattern, demonstrating that the inhibitory effect of the exocyst complex on WPB exocytosis can be separated from its role in WPB biogenesis.
Methods
Reagents and antibodies
Culture media, endothelial growth factor supplements, and antibiotics were obtained from Lonza. Lipofectamine and Dynabeads were obtained from Invitrogen. Thrombin, epinephrine, and Endosidin2 were obtained from Sigma-Aldrich. Rabbit polyclonal unlabeled and horseradish peroxidase–conjugated anti-human VWF were obtained from Dako. Rabbit polyclonal anti-HPS6, -HPS5, -EXOC2, -EXOC4, and -EXO70 were obtained from Proteintech. Mouse monoclonal anti-VWF (F8/86), -CD63 (TS63), -EEA1, anti-HA, and rabbit polyclonal ant-Rab11 were obtained from Invitrogen. Mouse monoclonal anti-sec8/EXOC4 was obtained from Millipore. Rabbit polyclonal anti-TGN46 was obtained from Novus. Rabbit monoclonal anti-Rab7 (D95F2) was obtained from CST. Species-specific horseradish peroxidase–conjugated secondary antibodies used for immunoblotting were obtained from Abcam. Specifies-specific Alexa Fluor 488 and 568 conjugated secondary antibodies were obtained from Invitrogen.
Human umbilical vein endothelial cell culture
Human umbilical vein endothelial cells (HUVECs; Lonza) were cultured as previously described.22 Only passages 2 to 4 were used.
siRNA and DNA constructs, transfection, and transduction
Predesigned HPS6 (s36366), HPS5 (s22172), EXOC4 (s334061), EXOC2 (s31470), EXO70 (s23432), and negative control (4390844) siRNAs were obtained from Invitrogen. siRNA transfection was performed using Lipofectamine RNAimax per the manufacturer’s protocol.
The lentiviral constructs pLKO.1 containing HPS6 shRNAs (clone IDs HsSH00178401 and HsSH00178412) and Phage-CMV-C-FLAG-HA-IRES-Puro containing human HPS6 mRNA (clone ID HsCD00459726) were obtained from the Harvard PlasmID Database. The mammalian expression vector CD63-pEGFP C2 was obtained from Addgene (#62964) and subcloned into a lentiviral construct pLX304 also obtained from Addgene using the pENTR/TOPO cloning kit (Thermo Fisher Scientific). Lentiviral particles were generated using a previously published protocol.
VWF assays
VWF Ag enzyme-linked immunosorbent assay (ELISA) and multimer assays were carried out as previously described.33
Immunoprecipitation and Western blotting
Immunoprecipitation (IP) was carried out on cytosolic fractions prepared using Tween-20 buffer using protein G Dynabeads (Invitrogen) following the manufacturer’s protocol. Western blots were carried out as previously described.22
Microscopy
Immunofluorescence microscopy (IF) was carried out on HUVECs plated on 1% gelatin-coated coverslips in 24-well plates, fixed with 4% paraformaldehyde, labeled with primary and secondary antibodies, mounted on Aqua poly/mount (Polysciences) using Olympus BX62 microscope equipped with a 100X Plan Apochromat objective lens and digital QImaging Rolera e-mc2 camera. Images were processed using SlideBook 6 software.
Electron microscopy (EM) protocol has been previously described.22
For super-resolution by radial fluctuation (SRRF) analysis, IF was carried out using a Nikon i90 Andor equipped with a 100x1.40 CFI Plan Apo VC Oil objective lens and iXon Ultra 888 Andor camera controlled by Micromanager. Super-resolution images were created from a stack of 500 frames recorded at 1-ms exposure time using NanoJ-SRRF with the GPU functionality in image acquisition. For SRRF reconstruction, radiality magnification was set to 5 (1 pixel = 21.4 nm), the ring radius was set to 0.73, and axes in the ring were set to 8.
For live-cell imaging, CD63-GFP HUVECs were plated on matrigel-coated 35-mm glass-bottom dishes and imaged on a Nikon Ti motorized inverted microscope equipped with an environmental chamber at 37 C/5% CO2, Yokagawa CSU-X1 spinning disk confocal, and Hamamatsu Flash 4.0 V3 sCMOS camera. Time-lapse microscopy was performed by capturing images over 5 minutes at a frame rate of approximately 3 fps using MetaMorph (Molecular Devices) software. Images were further processed in ImageJ software.
Mice
C57BL/6J mice and B6.Cg-Hps6ru/J mice were obtained from The Jackson Laboratory (Bar Harbor, ME). The Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee approved all animal care and procedures.
Human plasma
Citrated whole blood was obtained by venipuncture from a volunteer and a Hermansky-Pudlak syndrome subtype 3 patient with Institutional Review Board approval and in accordance with the Declaration of Helsinki. Plasma was prepared by centrifugation of whole blood at 1500g for 15 minutes.
Statistical analysis
Statistical analysis was performed using an unpaired Student t test: *P < .05; **P < .02; and ***P < .001.
Results
WPBs are immature and mislocalized on BLOC-2 depletion
To understand the role of BLOC-2 in WPB biogenesis, we first characterized the localization and morphology of WPBs in BLOC-2–depleted endothelium. BLOC-2 subunit HPS6-depleted HUVECs were generated by transduction of cells with lentiviral particles containing HPS6 shRNA. Two separate shRNA were used (supplemental Figure 1A, available on the Blood Web site). Control cells were generated in parallel by using empty vectors. Immunofluorescence microscopy analysis of HUVECs labeled with anti-VWF antibody revealed that the localization and structure of WPBs was significantly altered on BLOC-2 depletion (Figure 1A). WPBs are characteristically linear, rod-shaped structures, largely in the periphery of the cell. BLOC-2 depletion resulted in rounded and clumped WPBs, restricted to the perinuclear space. BLOC-2-depleted HUVECs had significantly lower number of mature WPBs compared with the controls (Figure 1B). Transmission EM analysis of BLOC-2–depleted HUVECs also revealed a lack of mature WPBs with only immature WPBs in the vicinity of the TGN (Figure 1C). Abnormal round membrane-bound structures more than 500 nm in diameter with unfolded membranous contents were notable in BLOC-2–depleted HUVECs (supplemental Figure 1B). Localization studies showed that immature WPBs in BLOC-2–depleted cells were present in close proximity to Rab11-positive endosomes (Figure 1D-E) in the vicinity of the TGN (supplemental Figure 1C-D). In contrast, the late endosome marker Rab7 did not associate with immature WPBs in BLOC-2–depleted cells (supplemental Figure 2A), whereas the early endosome marker EEA1 associated with WPBs more frequently in control cells (supplemental Figure 2B). Overall, although endosomal morphology is not markedly altered in BLOC-2–depleted HUVECs, WPBs are immature in the BLOC-2–depleted cells and show increased association with Rab11 but decreased association with EEA1.
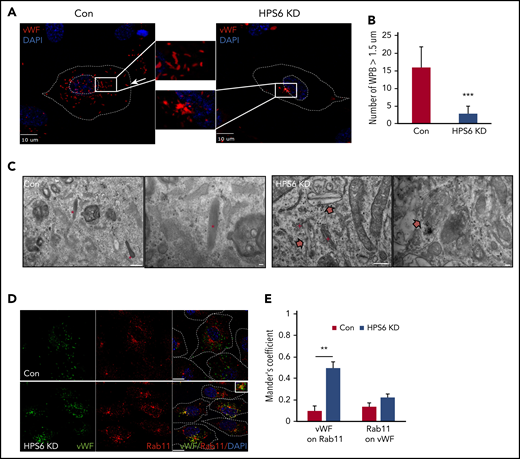
Depletion of BLOC-2 results in rounded, immature WPBs that are trapped in a post-TGN compartment. (A) IF analyses of BLOC-2 component HPS6-depleted and control HUVECs labeled with anti-VWF antibody and counterstained with DAPI. The insets are 4× magnifications of the boxed regions. Rod-shaped, mature WPBs distributed throughout the cell can be observed in control cells, but only round, immature WPBs, clumped perinuclearly, are evident in BLOC-2–depleted cells. Scale bars represent 10 μm. (B) Fifteen BLOC-2–depleted and control cells were randomly selected and total number of WPBs > 1.5 μM in length were determined for 2 independent studies (***P < .001). (C) Transmission EM analyses of BLOC-2–depleted and control HUVECs showing presence of mature, rod-shaped WPBs (*) in control cells with characteristic striated appearance, whereas BLOC-2–depleted cells lacked mature WPBs altogether. Instead, only immature WPBs (block arrows) in the vicinity of the Golgi apparatus (+) were observed. Scale bars represent 500 nm (long bar) and 100 nm (short bars). (D) IF analyses of BLOC-2–depleted and control HUVECs dual-labeled with anti-VWF and Rab11 antibodies and counterstained with DAPI. The inset (magnified 4×) highlights colabeling of VWF and Rab11 in BLOC-2–depleted cells. Scale bars represent 10 μm. (E) Bar graph showing Mander’s overlap coefficient of VWF staining on Rab11 and Rab11 on VWF in Con and BLOC-2–depleted cells. Fifteen BLOC-2–depleted and control cells each from 2 experiments were analyzed (**P < .01).
Depletion of BLOC-2 results in rounded, immature WPBs that are trapped in a post-TGN compartment. (A) IF analyses of BLOC-2 component HPS6-depleted and control HUVECs labeled with anti-VWF antibody and counterstained with DAPI. The insets are 4× magnifications of the boxed regions. Rod-shaped, mature WPBs distributed throughout the cell can be observed in control cells, but only round, immature WPBs, clumped perinuclearly, are evident in BLOC-2–depleted cells. Scale bars represent 10 μm. (B) Fifteen BLOC-2–depleted and control cells were randomly selected and total number of WPBs > 1.5 μM in length were determined for 2 independent studies (***P < .001). (C) Transmission EM analyses of BLOC-2–depleted and control HUVECs showing presence of mature, rod-shaped WPBs (*) in control cells with characteristic striated appearance, whereas BLOC-2–depleted cells lacked mature WPBs altogether. Instead, only immature WPBs (block arrows) in the vicinity of the Golgi apparatus (+) were observed. Scale bars represent 500 nm (long bar) and 100 nm (short bars). (D) IF analyses of BLOC-2–depleted and control HUVECs dual-labeled with anti-VWF and Rab11 antibodies and counterstained with DAPI. The inset (magnified 4×) highlights colabeling of VWF and Rab11 in BLOC-2–depleted cells. Scale bars represent 10 μm. (E) Bar graph showing Mander’s overlap coefficient of VWF staining on Rab11 and Rab11 on VWF in Con and BLOC-2–depleted cells. Fifteen BLOC-2–depleted and control cells each from 2 experiments were analyzed (**P < .01).
BLOC-2 mediates tubulovesicular transport from endosomes to maturing WPBs
As WPBs develop, they incorporate additional proteins essential for their maturation and eventual release.14 The multifunctional tetraspanin CD63 is delivered to the maturing WPBs via input from the endosomes.15,16 IF with dual labeling for VWF and CD63 demonstrated strong colocalization in mature WPBs in the periphery of control cells. However, BLOC-2–depleted HUVECs lacked CD63 labeling of peripheral WPBs (Figure 2A). There was colabeling of VWF and CD63 more so in immature WPBs in BLOC-2–depleted HUVECs than controls, perhaps secondary to the close proximity of immature WPBs to perinuclear CD63-containing endosomes (Figure 2A). Super-resolution microscopy showed that the signal originating from VWF was separate from CD63 in BLOC-2–depleted HUVECs, unlike mature WPBs in control HUVECs, confirming that CD63 does not localize to immature WPBs in BLOC-2–depleted HUVECS. To evaluate this further, we expressed GFP-tagged CD63 under the control of the CMV promoter in HUVECs using lentiviral transduction. IF revealed strong GFP signal in endosomal structures in these cells but also in distinct cigar-shaped WPBs distributed throughout the cell. Labeling of these cells with anti-VWF antibody confirmed the presence of VWF in these GFP-positive WPBs (supplemental Figure 2C). Furthermore, siRNA-mediated BLOC-2 component HPS6 depletion in these GFP-CD63–expressing HUVECs resulted in elimination of GFP labeling of WPBs seen in control cells at 48 hours (Figure 2B; supplemental Figure 2C), indicating that CD63 is not delivered to immature WPBs in BLOC-2–depleted cells.
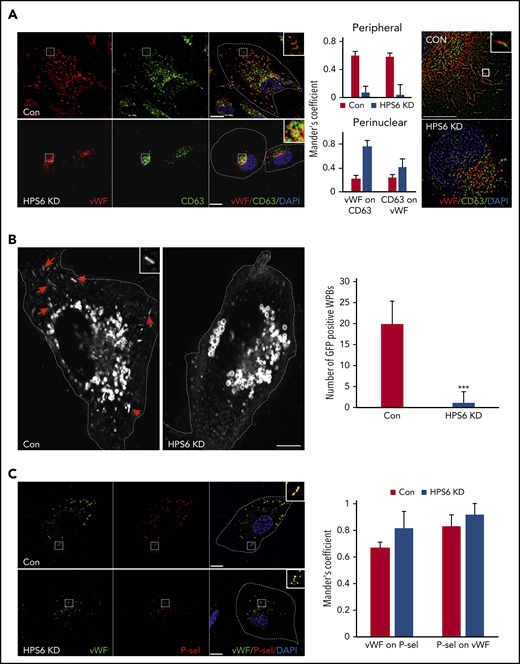
BLOC-2–depleted immature WPBs lack endosomal input. (A) IF analyses of BLOC-2–depleted and control HUVECs dual-labeled with anti-VWF and CD63 antibodies, counterstained with DAPI. The insets highlight colabeling of mature WPBs with CD63 in control cells in contrast to perinuclear localization of the 2 in BLOC-2–depleted cells. Scale bars represent 10 μm. The bar graphs show Mander’s overlap coefficient. Fifteen cells each from 2 experiments were analyzed. The right panel shows super-resolution analysis using radial fluctuation (SRRF) of anti-VWF and anti-CD63 dual-labeled HUVECs confirming separation of VWF and CD63 signals in the perinuclear immature WPBs in BLOC-2–depleted cells compared with colabeled peripheral mature WPBs in control cells. Scale bars represent 2.5 μm. (B) IF analysis of HUVECs transduced with GFP-CD63 expressing lentiviral particles 48 hours after transfection with either control or HPS6 siRNA. Arrows show GFP-labeled mature rod-shaped WPBs in control cells missing in BLOC-2–depleted cells and the inset highlights a mature WPB. GFP labels endosomes in both cell types. Scale bars represent 10 μm. Bar graph compares total number of GFP-labeled mature WPBs per cell in control compared with BLOC-2–depleted cells. Fifteen cells each from 2 experiments were analyzed (***P < .001). (C) IF analyses of BLOC-2–depleted and control HUVECs dual-labeled with anti-P-selectin and CD63 antibodies, counterstained with DAPI. The insets highlight colabeling of VWF and P-selectin in both control and BLOC-2–depleted cells. Scale bars represent 10 μm. The bar graphs show Mander’s overlap coefficient. Fifteen cells each from 2 experiments were analyzed.
BLOC-2–depleted immature WPBs lack endosomal input. (A) IF analyses of BLOC-2–depleted and control HUVECs dual-labeled with anti-VWF and CD63 antibodies, counterstained with DAPI. The insets highlight colabeling of mature WPBs with CD63 in control cells in contrast to perinuclear localization of the 2 in BLOC-2–depleted cells. Scale bars represent 10 μm. The bar graphs show Mander’s overlap coefficient. Fifteen cells each from 2 experiments were analyzed. The right panel shows super-resolution analysis using radial fluctuation (SRRF) of anti-VWF and anti-CD63 dual-labeled HUVECs confirming separation of VWF and CD63 signals in the perinuclear immature WPBs in BLOC-2–depleted cells compared with colabeled peripheral mature WPBs in control cells. Scale bars represent 2.5 μm. (B) IF analysis of HUVECs transduced with GFP-CD63 expressing lentiviral particles 48 hours after transfection with either control or HPS6 siRNA. Arrows show GFP-labeled mature rod-shaped WPBs in control cells missing in BLOC-2–depleted cells and the inset highlights a mature WPB. GFP labels endosomes in both cell types. Scale bars represent 10 μm. Bar graph compares total number of GFP-labeled mature WPBs per cell in control compared with BLOC-2–depleted cells. Fifteen cells each from 2 experiments were analyzed (***P < .001). (C) IF analyses of BLOC-2–depleted and control HUVECs dual-labeled with anti-P-selectin and CD63 antibodies, counterstained with DAPI. The insets highlight colabeling of VWF and P-selectin in both control and BLOC-2–depleted cells. Scale bars represent 10 μm. The bar graphs show Mander’s overlap coefficient. Fifteen cells each from 2 experiments were analyzed.
To compare this finding to a cargo of the synthetic pathway of WPB biogenesis, we evaluated localization of P-selectin, an integral WPB transmembrane protein.34,35 Unlike CD63, P-selectin trafficked to nascent WPBs by its direct association with VWF in TGN.36 IF with dual labeling for P-selectin and VWF revealed that immature WPBs in BLOC-2–depleted HUVEC labeled with P-selectin (Figure 2C). Absence of CD63 but not P-selectin indicates that BLOC-2 regulates trafficking of endosomal cargo to maturing WPBs after synthesis at TGN.
Next, we performed live-cell microscopy on GFP-CD63 expressing HUVECs to study interactions between endosomes and maturing WPBs. Because GFP-CD63 localizes to both endosomes and WPBs, it enabled visualization of both organelles and their interactions over time. Frequent GFP-CD63–positive elements, likely transport carriers, were identified exiting endosomes. Many were vesicular in nature, whereas others were tubular (Figure 3A). Long endosomal tubular extensions terminating on WPBs were frequently evident (Figure 3B-C; supplemental Video 1). The number of these endosomal tubular extensions were not altered in BLOC-2–depleted GFP-CD63 HUVECs but were misdirected to the core of the cell, perhaps toward the TGN (Figure 3B-C; supplemental Video 2). Such misdirection of endosomal tubular extensions has been previously noted in melanocytes.23 Direct visualization of CD63 carrying endosomal transport tubules directed toward WPBs and loss of CD63 expression on WPBs in BLOC-2–depleted cells confirms the essential role that BLOC-2 plays in WPB maturation.
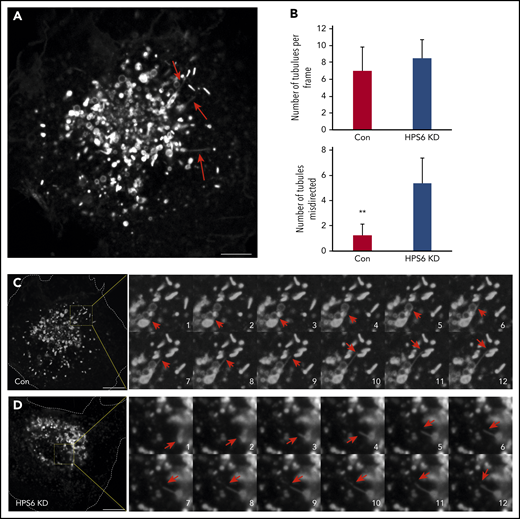
Live cell imaging shows misdirection of endosome-derived transport tubules in endothelial cells lacking BLOC-2. (A) GFP-CD63 expressing HUVECs were analyzed by live-cell spinning-disc confocal microscopy and demonstrate numerous endosome-derived long transport tubules (arrows) and GFP-CD63-labeled mature WPBs. Scale bar represents 5 μm. (B) Bar graphs showing total number of long transport tubules exiting endosomes per cell estimated by analyzing first frame from 8 cells each from 2 separate experiments, and number of tubules directed to the core of the cell in control and BLOC-2–depleted GFP-CD63-expressing HUVECs (**P < .01). (C) Fourfold magnified 0.3-s interval image sequences (1 to 12) are shown from the boxed region in the micrograph of a control cell on the left. A GFP-CD63–positive long transport tubule (red arrows) exits an endosome and ends on a WPB. Scale bar represents 10 μm. (D) Fourfold magnified 0.3-s interval image sequences (1 to 12) are shown from the boxed region in the micrograph of a BLOC-2–depleted cell on the left. A GFP-CD63–positive long transport tubule (red arrows) exits an endosome but is directed centrally to another endosome. Scale bar represents 10 μm.
Live cell imaging shows misdirection of endosome-derived transport tubules in endothelial cells lacking BLOC-2. (A) GFP-CD63 expressing HUVECs were analyzed by live-cell spinning-disc confocal microscopy and demonstrate numerous endosome-derived long transport tubules (arrows) and GFP-CD63-labeled mature WPBs. Scale bar represents 5 μm. (B) Bar graphs showing total number of long transport tubules exiting endosomes per cell estimated by analyzing first frame from 8 cells each from 2 separate experiments, and number of tubules directed to the core of the cell in control and BLOC-2–depleted GFP-CD63-expressing HUVECs (**P < .01). (C) Fourfold magnified 0.3-s interval image sequences (1 to 12) are shown from the boxed region in the micrograph of a control cell on the left. A GFP-CD63–positive long transport tubule (red arrows) exits an endosome and ends on a WPB. Scale bar represents 10 μm. (D) Fourfold magnified 0.3-s interval image sequences (1 to 12) are shown from the boxed region in the micrograph of a BLOC-2–depleted cell on the left. A GFP-CD63–positive long transport tubule (red arrows) exits an endosome but is directed centrally to another endosome. Scale bar represents 10 μm.
BLOC-2 interacts with the exocyst complex
To identify binding partners of BLOC-2 in endothelium, IP of endogenous BLOC-2 protein subunits HPS5 and HPS6 were performed in HUVEC lysates separately and co-IP proteins were analyzed by mass spectrometry. IP with host-specific polyclonal IgG was performed in parallel. A list of proteins that were pulled down exclusively with anti-HPS6 and -HPS5 antibodies but absent in IgG control was then generated (supplemental Table). Among these, 3 subunits of the exocyst complex, EXOC2 (sec5), EXOC4 (sec8), and EXO70, were enriched in both HPS6 and HPS5 co-IPs. Given that BLOC-2 had previously been thought to function as a tether,23 its interaction with a tethering complex seemed plausible. This interaction was also observed by co-IP and Western blot analysis. Furthermore, reverse co-IP of HUVEC lysates performed with anti-EXOC4 antibody demonstrated EXOC4 binding to BLOC-2 (Figure 4A). To confirm this interaction, we transduced HUVECs with lentiviral particles containing HA-tagged HPS6 under the control of CMV promoter or empty vector. IP of HA-HPS6 or control cells performed using anti-HA antibody confirmed the presence of EXOC4 in co-IP of HA-HPS6-expressing cells (supplemental Figure 3A). Mass spectrometry of IP proteins enriched for other BLOC-2 subcomponents, HPS3 and HPS5, suggesting that overexpressed HA-HPS6 forms BLOC-2 complex as in its native state and immune-electron microscopy revealed presence of HPS6 on the limiting membrane of CD63-positive endosomes (supplemental Figure 3B). To evaluate the specificity of BLOC-2 and EXOC4 interaction, IPs with anti-HPS1, a component of BLOC-3, anti-AP3B1 and anti–myosin-actin crosslinking factor 1 (MACF1) were performed. EXOC4 did not co-IP with these antibodies (supplemental Figure 3C-E). In addition to its role in exocytosis, exocyst is found in mammalian endosomes where it is involved in vesicular trafficking.25,29 IF with labeling for EXOC4 showed abundant perinuclear localization of exocyst in HUVECs with partial colabeling with CD63 (Figure S3F).
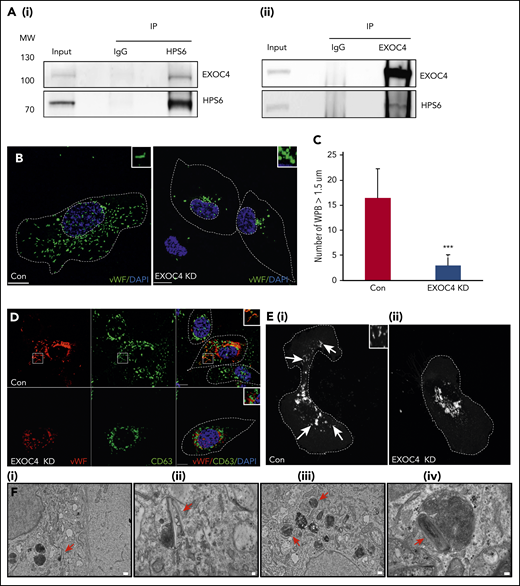
BLOC-2 interacts with the exocyst complex, which is essential for WPB biogenesis. (A) IPs of HUVEC lysates with anti-HPS6 (i) or EXOC4 (ii) antibodies resolved by SDS-PAGE and immunoblotted with HPS6 and EXOC4 antibodies. HUVEC lysate (∼5% of IP) used as input and IP with polyclonal IgG used as a control for both immunoblots. (B) IF analyses of EXOC4-depleted and control HUVECs labeled with anti-VWF antibody and counterstained with DAPI. As for BLOC-2–depleted cells, only rounded, immature WPBs, which are clumped perinuclearly are evident, highlighted by insets. Scale bars represent 10 μm. (C) Fifteen BLOC-2–depleted and control cells were randomly selected and total number of WPBs > 1.5 μM in length were determined for 2 independent studies (***P < .001). (D) EXOC4-depleted and control HUVECs dual labeled with anti-VWF and CD63 antibodies were analyzed by IF, counterstained with DAPI. The insets highlight colabeling of mature WPBs and CD63 in control cells, absent in EXOC4-depleted cells. Scale bars represent 10 μm. (E) GFP-CD63–expressing HUVECs analyzed by IF 48 hours after transfection with control or EXOC4 siRNA. Arrows show GFP-CD63 labeling of mature WPBs in control cells (i), absent in EXOC4-depleted cells (ii). Inset highlights mature WPBs. Scale bars represent 10 μm. (F) EM analyses of EXOC4-depleted HUVECs show immature WPBs with unfurled VWF (arrows). Scale bars represent 500 nm (i and iii) and 100 nm (ii and iv).
BLOC-2 interacts with the exocyst complex, which is essential for WPB biogenesis. (A) IPs of HUVEC lysates with anti-HPS6 (i) or EXOC4 (ii) antibodies resolved by SDS-PAGE and immunoblotted with HPS6 and EXOC4 antibodies. HUVEC lysate (∼5% of IP) used as input and IP with polyclonal IgG used as a control for both immunoblots. (B) IF analyses of EXOC4-depleted and control HUVECs labeled with anti-VWF antibody and counterstained with DAPI. As for BLOC-2–depleted cells, only rounded, immature WPBs, which are clumped perinuclearly are evident, highlighted by insets. Scale bars represent 10 μm. (C) Fifteen BLOC-2–depleted and control cells were randomly selected and total number of WPBs > 1.5 μM in length were determined for 2 independent studies (***P < .001). (D) EXOC4-depleted and control HUVECs dual labeled with anti-VWF and CD63 antibodies were analyzed by IF, counterstained with DAPI. The insets highlight colabeling of mature WPBs and CD63 in control cells, absent in EXOC4-depleted cells. Scale bars represent 10 μm. (E) GFP-CD63–expressing HUVECs analyzed by IF 48 hours after transfection with control or EXOC4 siRNA. Arrows show GFP-CD63 labeling of mature WPBs in control cells (i), absent in EXOC4-depleted cells (ii). Inset highlights mature WPBs. Scale bars represent 10 μm. (F) EM analyses of EXOC4-depleted HUVECs show immature WPBs with unfurled VWF (arrows). Scale bars represent 500 nm (i and iii) and 100 nm (ii and iv).
Exocyst depletion impairs WPB maturation
We next evaluated the effect of the exocyst complex depletion on WPB maturation. Depletion of any 1 subunit of this obligate complex disrupts its function.37,38 Furthermore, the octameric complex is built of 2 subcomplexes: I and II. Subcomplex I is comprised of subunits EXOC1, EXOC2, EXOC3, and EXOC4, whereas subcomplex II is comprised of subunits EXOC5, EXOC6, EXO70, and EXO84. EXOC4 depletion in HUVECs resulted in reduction of EXOC2 but not of subcomplex II subunit EXO70 (supplemental Figure 4A).37,38 Similarly, EXO70 depletion did not reduce subcomplex I subunit EXOC4 levels (supplemental Figure 4B), suggesting similar subcomplex organization in endothelial cells. Deficiency of EXOC4 resulted in a phenotype similar to BLOC-2 depletion with significant reduction in mature WPBs (Figure 4B-C). EXOC4-depleted HUVECs labeled with anti-VWF antibody showed clumped and rounded WPBs located perinuclearly, lacking CD63 labeling of mature WPBs (Figure 4D). There was partial colabeling of VWF and CD63 perinuclearly, more in EXOC4-depleted cells, but also in controls. EXOC4 depletion of GFP-CD63 HUVECs also resulted in loss of GFP-CD63 labeling of mature WPBs (Figure 4E). EM analysis showed lack of mature WPBs but presence of immature WPBs in EXOC4-depleted HUVECs (Figure 4F). Overall, disruption of WPB maturation in exocyst-depleted cells resembled that observed in BLOC-2–depleted cells (Figures 1 and 2).
BLOC-2 depletion impairs VWF multimerization and WPB exocytosis
We next evaluated the impact of BLOC-2 depletion on VWF exocytosis and multimerization. In HUVECs transduced with lentiviral particles containing HPS6 shRNA, thrombin-induced VWF exocytosis was significantly impaired (37 ± 3% compared with control at 30 minutes; Figure 5A-B). The total VWF antigen in BLOC-2–depleted HUVECs was not reduced, implying VWF synthesis was not affected (Figure 5C). Basal VWF, estimated as the total VWF antigen in media collected after overnight incubation without any chemical agonist, was also reduced (59 ± 4% compared with control; Figure 5D). Depletion of BLOC-2 subunit HPS5 using siRNA also impaired thrombin-induced VWF exocytosis (Figure S5A). Additionally, depletion of BLOC-2 subunit HPS6 in primary human dermal microvasculature endothelial cells and EAhy.926, an endothelial cell line, impaired thrombin-induced VWF exocytosis (supplemental Figure 5B-C).
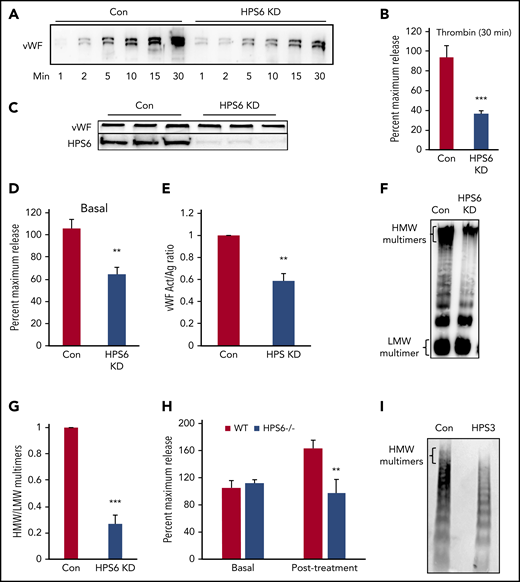
BLOC-2 depletion impairs endothelial WPB exocytosis and VWF multimerization. (A) Immunoblot showing total VWF antigen levels in the media at 1, 2, 5, 10, 15, and 30 minutes after 1 U/mL thrombin stimulation of BLOC-2–depleted and control HUVECs resolved by SDS-PAGE and immunoblotted with anti-VWF antibody. (B) VWF antigen released from HPS6-depleted and control HUVECs 30 minutes after 1 U/mL thrombin stimulation determined by ELISA. (***P < .001; n = 3). (C) Whole cell lysates of BLOC-2–depleted and control cells were resolved by SDS-PAGE and immunoblotted with antibodies to VWF and HPS6. Total endothelial cell VWF content is unaltered on BLOC-2 depletion. (D) Resting conditioned media was collected after 8-hour incubation with control or BLOC-2–depleted HUVECs. VWF antigen levels were measured using VWF ELISA (**P < .01; n = 3). (E) BLOC-2–depleted and control HUVECs were stimulated with 1 U/mL thrombin, and media were collected at 30 minutes. Total VWF antigen (Ag) and activity (Act) were measured using a VWF ELISA and a type 3 collagen binding assay, respectively. The bar graph shows the Act to Ag ratios (**P < .01; n = 3). (F) Qualitative analysis of exocytosed VWF multimers was performed using agarose gel electrophoresis. BLOC-2–depleted and control HUVECs were stimulated with 1 U/mL thrombin. Harvested media concentrated in centrifugal concentrators and fractionated by nonreducing 1.4% agarose gel electrophoresis was immunoblotted with anti-VWF antibodies. Blot shows reduced HMW multimers in BLOC-2–depleted cells. (G) Densitometry analysis of VWF multimers in e quantified as the ratio of HMW multimer to the lowest-molecular-weight (LMW) oligomer, as shown. (n = 3; ***P < .001). (H) WT and HPS6−/− mice were treated subcutaneously with epinephrine. VWF antigen levels in plasma obtained before and 15 minutes after epinephrine treatment were determined using VWF ELISA (n = 3; **P < .01). (I) Plasma from an individual with a BLOC-2 mutation and a healthy control was resolved by nonreducing 1.4% agarose gel electrophoresis and immunoblotted with VWF antibody showing reduced HMW multimers.
BLOC-2 depletion impairs endothelial WPB exocytosis and VWF multimerization. (A) Immunoblot showing total VWF antigen levels in the media at 1, 2, 5, 10, 15, and 30 minutes after 1 U/mL thrombin stimulation of BLOC-2–depleted and control HUVECs resolved by SDS-PAGE and immunoblotted with anti-VWF antibody. (B) VWF antigen released from HPS6-depleted and control HUVECs 30 minutes after 1 U/mL thrombin stimulation determined by ELISA. (***P < .001; n = 3). (C) Whole cell lysates of BLOC-2–depleted and control cells were resolved by SDS-PAGE and immunoblotted with antibodies to VWF and HPS6. Total endothelial cell VWF content is unaltered on BLOC-2 depletion. (D) Resting conditioned media was collected after 8-hour incubation with control or BLOC-2–depleted HUVECs. VWF antigen levels were measured using VWF ELISA (**P < .01; n = 3). (E) BLOC-2–depleted and control HUVECs were stimulated with 1 U/mL thrombin, and media were collected at 30 minutes. Total VWF antigen (Ag) and activity (Act) were measured using a VWF ELISA and a type 3 collagen binding assay, respectively. The bar graph shows the Act to Ag ratios (**P < .01; n = 3). (F) Qualitative analysis of exocytosed VWF multimers was performed using agarose gel electrophoresis. BLOC-2–depleted and control HUVECs were stimulated with 1 U/mL thrombin. Harvested media concentrated in centrifugal concentrators and fractionated by nonreducing 1.4% agarose gel electrophoresis was immunoblotted with anti-VWF antibodies. Blot shows reduced HMW multimers in BLOC-2–depleted cells. (G) Densitometry analysis of VWF multimers in e quantified as the ratio of HMW multimer to the lowest-molecular-weight (LMW) oligomer, as shown. (n = 3; ***P < .001). (H) WT and HPS6−/− mice were treated subcutaneously with epinephrine. VWF antigen levels in plasma obtained before and 15 minutes after epinephrine treatment were determined using VWF ELISA (n = 3; **P < .01). (I) Plasma from an individual with a BLOC-2 mutation and a healthy control was resolved by nonreducing 1.4% agarose gel electrophoresis and immunoblotted with VWF antibody showing reduced HMW multimers.
We determined the functional activity of exocytosed VWF from BLOC-2–depleted HUVECs by assaying its collagen binding activity, as previously described.39 Type 3 collagen binding activity of VWF, normalized to the amount of total VWF released, was significantly reduced in BLOC-2–depleted HUVECs compared with controls (Figure 5E). Because the functional activity of VWF is directly proportional to the presence of HMW multimers, the multimeric state of the exocytosed VWF was determined.1 Thrombin-induced HUVEC releasates were prepared and concentrated ∼100-fold using centrifugal filters. Concentrated media were then subjected to 1.4% agarose gel electrophoresis in nonreducing conditions and immunoblotted for VWF. BLOC-2 depletion resulted in deficiency of HMW multimers (Figure 5F-G).
HPS6−/− mice were next studied using a model of epinephrine-induced VWF release.40 Basal and post–epinephrine treatment plasma samples were obtained in HPS6−/− and WT mice, and VWF antigen was determined. VWF antigen increased to 158 ± 12% compared with basal levels after systemic administration of epinephrine in WT mice compared with 93 ± 20% of basal in HPS6−/− mice (Figure 5H). Plasma VWF multimers were also evaluated in a BLOC2-deficient individual with normal plasma VWF antigen levels. Compared with a healthy control, HMW multimers were reduced (Figure 5I).
Exocyst depletion impairs VWF multimerization but augments WPB exocytosis
We next evaluated the impact of exocyst depletion on VWF exocytosis and multimerization. Exocyst regulates granule exocytosis in many cell types but has not been evaluated in endothelium.25,37,41-44 Unexpectedly, thrombin-induced VWF exocytosis was significantly augmented in EXOC4-depleted HUVECs (207 ± 7%) compared with controls (Figure 6A-B). There was no increase in basal release (Figure 6C). We then evaluated the multimeric state of VWF exocytosed from EXOC4-depleted HUVECs as above. Despite increased VWF release, EXOC4 depletion resulted in deficiency of HMW multimers (Figure 6D-E). Concurrent depletion of both BLOC-2 and EXOC4 significantly increased VWF exocytosis compared with BLOC-2 depletion alone (Figure 6F). Depletion of BLOC-2 subunit HPS6 did not alter EXOC4 levels in HUVECs (supplemental Figure 5D).
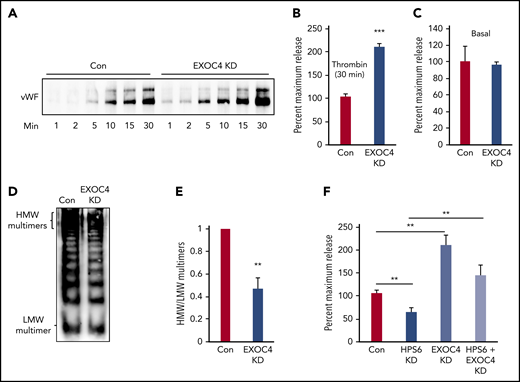
Exocyst depletion impairs VWF multimerization but augments WPB exocytosis. (A) Immunoblot showing total VWF antigen levels in media of EXOC4-depleted and control HUVECs at 1, 2, 5, 10, 15, and 30 minutes after stimulation with 1 U/mL thrombin resolved by SDS-PAGE and immunoblotted with anti-VWF antibody. (B) Densitometry of 3 individual thrombin-stimulation experiments as described in panel A showing significant augmentation of VWF exocytosis on EXOC4 depletion at 30 minutes (***P < .001). (C) Resting conditioned media was collected following 8-hour incubation with control or EXOC4-depleted HUVECs. VWF antigen levels were measured using VWF ELISA. (D) EXOC4-depleted and control HUVECs were stimulated with 1 U/mL thrombin. Harvested media concentrated in centrifugal concentrators and fractionated by nonreducing 1.4% agarose gel electrophoresis was immunoblotted using anti-VWF antibodies. Blot shows reduced HMW multimers in EXOC4-depleted cells. (E) Densitometry analysis of VWF multimers in panel D quantified as the ratio of HMW multimer to the LMW oligomer. (n = 2; **P < .01). (F) Control and BLOC-2–, EXOC4–, and combined BLOC-2– and EXOC4–depleted cells were stimulated with 1 U/mL thrombin, and total VWF antigen in media at 30 minutes was measured using ELISA. The bar graph shows that differing from BLOC-2 depletion, EXOC4 depletion augments VWF exocytosis. EXOC4 depletion augments VWF exocytosis in BLOC-2–depleted cells compared with BLOC-2 depletion alone (**P < .001 from 2 individual experiments each with triplicates).
Exocyst depletion impairs VWF multimerization but augments WPB exocytosis. (A) Immunoblot showing total VWF antigen levels in media of EXOC4-depleted and control HUVECs at 1, 2, 5, 10, 15, and 30 minutes after stimulation with 1 U/mL thrombin resolved by SDS-PAGE and immunoblotted with anti-VWF antibody. (B) Densitometry of 3 individual thrombin-stimulation experiments as described in panel A showing significant augmentation of VWF exocytosis on EXOC4 depletion at 30 minutes (***P < .001). (C) Resting conditioned media was collected following 8-hour incubation with control or EXOC4-depleted HUVECs. VWF antigen levels were measured using VWF ELISA. (D) EXOC4-depleted and control HUVECs were stimulated with 1 U/mL thrombin. Harvested media concentrated in centrifugal concentrators and fractionated by nonreducing 1.4% agarose gel electrophoresis was immunoblotted using anti-VWF antibodies. Blot shows reduced HMW multimers in EXOC4-depleted cells. (E) Densitometry analysis of VWF multimers in panel D quantified as the ratio of HMW multimer to the LMW oligomer. (n = 2; **P < .01). (F) Control and BLOC-2–, EXOC4–, and combined BLOC-2– and EXOC4–depleted cells were stimulated with 1 U/mL thrombin, and total VWF antigen in media at 30 minutes was measured using ELISA. The bar graph shows that differing from BLOC-2 depletion, EXOC4 depletion augments VWF exocytosis. EXOC4 depletion augments VWF exocytosis in BLOC-2–depleted cells compared with BLOC-2 depletion alone (**P < .001 from 2 individual experiments each with triplicates).
Regulatory role of exocyst on WPB biogenesis and exocytosis can be separated
Differential effects of BLOC-2 and EXOC4 depletion on WPB exocytosis led us to hypothesize that the exocyst complex, which plays an important role in granule exocytosis in other cell types,25 must serve a separate role in regulating WPB exocytosis that is independent of its role in endosomal trafficking. To evaluate this premise, we targeted exocyst component EXO70, which was also enriched in our HPS5 and HPS6 co-IPs, with the specific small molecular inhibitor Endosidin2.45 The goal was to inhibit exocyst complex rapidly without the need for depleting one or more of its components using RNAi. EXO70 depletion augmented thrombin-induced VWF exocytosis to a similar extent as EXOC4 depletion (199 ± 22% compared with control) at 48 hours after RNAi (Figure 7A). In contrast, Endosidin2 increased VWF release (192 ± 32% compared with control) over vehicle-treated controls at 2 hours, an incubation period insufficient to affect WPB biogenesis (Figure 7A). Consequently, releasates from Endosidin2-treated HUVECs contained HMW multimers (Figure 7B-C), unlike releasates from EXOC4- or EXO-70-depleted HUVECs. Endosidin2 treatment of BLOC-2-depleted cells increased VWF exocytosis in a dose-dependent manner (Figure 7D). The effect of Endosidin2 on thrombin-induced VWF exocytosis was readily reversible after a wash (Figure 7E). These findings confirm that regulatory role of exocyst in WPB biogenesis and exocytosis can be separated.
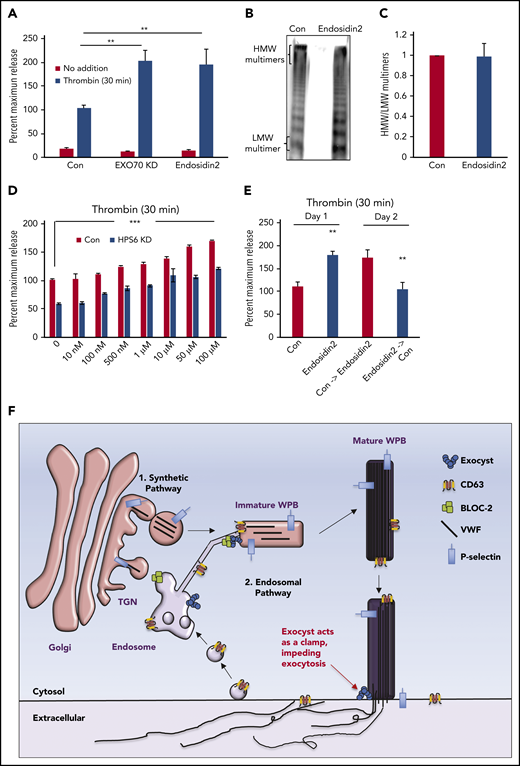
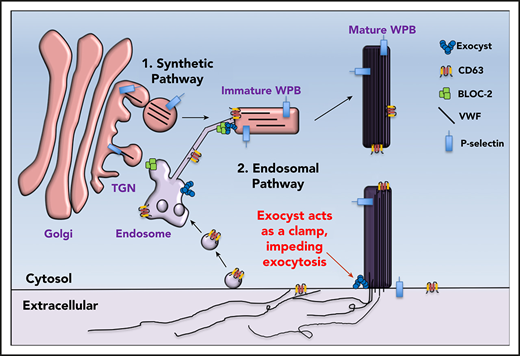
Regulatory functions of the exocyst complex on WPB maturation and WPB exocytosis can be separated. (A) Total VWF antigen was measured using ELISA in basal media (3 hours) and 30 minutes after thrombin stimulation (1 U/mL) in control, EXO70-depleted, and Endosidin2-treated (10 µM for 2 hours) HUVECs (**P < .01 from 3 individual experiments). (B) Qualitative VWF multimer analysis of concentrated media from thrombin-stimulated control or Endosidin2-treated HUVECs by nonreducing agarose gel electrophoresis and immunoblotting with anti-VWF antibodies show similar HMW multimers in control and Endosidin2-treated cells. (C) Densitometry analysis of VWF multimers in panel B quantified as the ratio of HMW multimer to the LMW oligomer (n = 3). (D) Dose-response analysis of Endosidin2 on thrombin-stimulated VWF release from control and BLOC-2–depleted HUVECs. BLOC-2–depleted HUVECs were treated with varying concentrations of Endosidin2 or DMSO for 2 hours and then stimulated with 1 U/mL thrombin, and total VWF antigen in media at 30 minutes was measured using ELISA (***P < .01 from 3 individual experiments). (E) Control and Endosidin2-treated (10 µM for 2 hours) HUVECs were stimulated with 1 U/mL thrombin, and media were collected at 30 minutes (day 1). After a wash, cells were reincubated with complete growth media. Twenty-four hours after initial treatment, controls from the previous day were exposed to Endosidin2 (10 µM for 2 hours) and originally Endosidin2-treated HUVECs were treated as controls. Cells were stimulated with 1 U/mL thrombin, and media were collected at 30 minutes (day 2). Total VWF antigen was measured in samples from both days using ELISA. (**P < .01 from 2 individual experiments each with triplicates). (F) Proposed model for BLOC-2 and exocyst function in endosomal transport during WPB biogenesis and VWF exocytosis. VWF is synthesized by endothelial cells and stored in WPBs. WPBs arise from direct tubulation of TGN membrane by newly synthesized VWF filaments. P-selectin enters nascent WPBs in direct association with VWF. Further maturation of WPBs continues as it relocates to the periphery of the cell. During this process, immature WPBs receive other cargoes essential for their maturation and develop HMW forms of VWF. One source of cargo is the endosomes. Cargo-carrying tubules exit endosomes and merge with maturing WPBs to deliver cargo such as CD63. This process is regulated by BLOC-2 that directly interacts with the exocyst complex to direct tubulovesicular transport from endosomes to WPBs. BLOC-2 depletion impairs exocytosis, but whether BLOC-2 promotes WPB release via a direct role in exocytosis is not known. Exocyst separately regulates WPB exocytosis at the plasma membrane, where, acting as a clamp, it inhibits exocytosis of mature WPBs. Release from the exocyst clamp augments release of VWF, including HMW multimers.
Regulatory functions of the exocyst complex on WPB maturation and WPB exocytosis can be separated. (A) Total VWF antigen was measured using ELISA in basal media (3 hours) and 30 minutes after thrombin stimulation (1 U/mL) in control, EXO70-depleted, and Endosidin2-treated (10 µM for 2 hours) HUVECs (**P < .01 from 3 individual experiments). (B) Qualitative VWF multimer analysis of concentrated media from thrombin-stimulated control or Endosidin2-treated HUVECs by nonreducing agarose gel electrophoresis and immunoblotting with anti-VWF antibodies show similar HMW multimers in control and Endosidin2-treated cells. (C) Densitometry analysis of VWF multimers in panel B quantified as the ratio of HMW multimer to the LMW oligomer (n = 3). (D) Dose-response analysis of Endosidin2 on thrombin-stimulated VWF release from control and BLOC-2–depleted HUVECs. BLOC-2–depleted HUVECs were treated with varying concentrations of Endosidin2 or DMSO for 2 hours and then stimulated with 1 U/mL thrombin, and total VWF antigen in media at 30 minutes was measured using ELISA (***P < .01 from 3 individual experiments). (E) Control and Endosidin2-treated (10 µM for 2 hours) HUVECs were stimulated with 1 U/mL thrombin, and media were collected at 30 minutes (day 1). After a wash, cells were reincubated with complete growth media. Twenty-four hours after initial treatment, controls from the previous day were exposed to Endosidin2 (10 µM for 2 hours) and originally Endosidin2-treated HUVECs were treated as controls. Cells were stimulated with 1 U/mL thrombin, and media were collected at 30 minutes (day 2). Total VWF antigen was measured in samples from both days using ELISA. (**P < .01 from 2 individual experiments each with triplicates). (F) Proposed model for BLOC-2 and exocyst function in endosomal transport during WPB biogenesis and VWF exocytosis. VWF is synthesized by endothelial cells and stored in WPBs. WPBs arise from direct tubulation of TGN membrane by newly synthesized VWF filaments. P-selectin enters nascent WPBs in direct association with VWF. Further maturation of WPBs continues as it relocates to the periphery of the cell. During this process, immature WPBs receive other cargoes essential for their maturation and develop HMW forms of VWF. One source of cargo is the endosomes. Cargo-carrying tubules exit endosomes and merge with maturing WPBs to deliver cargo such as CD63. This process is regulated by BLOC-2 that directly interacts with the exocyst complex to direct tubulovesicular transport from endosomes to WPBs. BLOC-2 depletion impairs exocytosis, but whether BLOC-2 promotes WPB release via a direct role in exocytosis is not known. Exocyst separately regulates WPB exocytosis at the plasma membrane, where, acting as a clamp, it inhibits exocytosis of mature WPBs. Release from the exocyst clamp augments release of VWF, including HMW multimers.
Discussion
In evaluating the role of BLOC-2 in WPB formation, we identified a previously unappreciated role of the exocyst complex in WPB maturation and VWF exocytosis. BLOC-2 binds exocyst and knockdown of either complex impairs delivery of endosomal components to developing WPBs resulting in the formation of rounded, clumped WPBs. Yet, in contrast to their similar and perhaps cooperative roles in WPB development, depletion of BLOC-2 and exocyst have opposite effects on WPB exocytosis. BLOC-2–deficient immature WPBs exhibit defective exocytosis, whereas depletion of exocyst augments exocytosis. Our studies show that, separate from its role in WPB biogenesis, inhibition of exocyst reversibly augments WPB exocytosis without altering VWF maturation, suggesting that exocyst serves as a clamp in the setting of exocytosis.
WPBs derive essential cargo from the endosomal compartment to mature, similar to other LROs.15-17 However, the role of protein complexes that are essential for formation of LROs and are defective in many known subtypes of HPS are poorly characterized in endothelial cells. We find that BLOC-2 directs endosome-derived tubular transport carriers to maturing WPBs. BLOC-2–deficient WPBs lacked tetraspanin CD63, an endosomal cargo, but not P-selectin, which enters WPBs directly bound to VWF filaments at the TGN.16 This confirms that in BLOC-2 deficiency the maturation of WPBs is impaired at a point after their initial formation at the TGN. In addition to CD63, we also find that EEA1, an early endosome marker, is localized to mature WPBs, but not immature WPBs in BLOC-2 deficient cells and may therefore be an endosomal cargo regulated by BLOC-2. Endosomal transport is also believed to be important for acidification of WPBs.19 Live cell microscopy of HUVECs expressing GFP-CD63 revealed frequent contacts between endosomes and WPBs via tubular extensions. Depletion of BLOC-2 did not alter endosomal expression of GFP-CD63 but eliminated the presence of GFP-CD63 on WPBs. GFP-CD63-positive tubular transport carriers exiting endosomes were present in BLOC-2–depleted cells but appeared misdirected, indicating that BLOC-2 directs endosome-derived tubular transport carriers to maturing WPBs. This phenomenon has been previously noted in melanosomes where deficiency of BLOC-2 redirects endosome-derived transport tubules to TGN or plasma membrane.23 BLOC-2 had been speculated to function as a tether in this role in melanosomes. Here, we find that BLOC-2 associates with a bona fide tether, the exocyst complex, regulating endosomal transport to maturing WPBs in endothelium.
Our proteomic analysis of immunoprecipitates generated using anti-HPS5 and -HPS6 antibodies enriched exocyst complex subunits EXOC2, EXOC4 and EXO70. The exocyst complex performs multiple essential intracellular functions.25 Among these, the role of exocyst in tethering secretory granules to plasma membrane preceding fusion and exocytosis in yeast has been most widely studied.25,26,38,41,44 Exocyst is also being increasingly placed in mammalian endosomes and endolysosomal trafficking pathways. Exocyst subunits EXOC4 and EXO70 were detected in Rab11-bearing endosomes that interacted and fused with phagosomes containing Staphylococcus aureus in endothelial cells, and depletion of these subunits greatly reduced acidification of phagosomes and elimination of phagocytosed Staphylococci.30,32 Depletion of EXO70 significantly increased exocytosis of the lysosomal enzyme β-hexosaminidase.31 We find exocyst to be present on CD63-positive endosomes in the endothelial cells, similar to BLOC-2. IF and EM confirm partial colocalization of EXOC4 with BLOC-2 subunit HPS6, specifically in small vesicular structures in close proximity to WPBs, which likely represent transport intermediates. Moreover, depletion of EXOC4 renders a phenotype similar to that of BLOC-2 depletion manifested by immature, rounded, WPBs that lack CD63, both by IF and EM. Interestingly, single nucleotide polymorphisms in EXOC2, EXOC4, and other exocyst complex subunits have been associated with variation in skin, hair, and eye pigmentation, as well as impaired platelet aggregation, the 2 classic manifestations of HPS, in genome-wide association studies.46-48
Despite their apparent physical association, BLOC-2 and exocyst have different functions in activation-dependent VWF release. Depletion of BLOC-2 inhibits thrombin-induced WPB release from HUVEC and impairs secretagogue-induced VWF exocytosis in BLOC-2−/− mice. Our data suggest that this defect in WPB exocytosis is linked to a WPB maturation defect, as is recognized for many other known components of WPB trafficking machinery.10,13,49,50 VWF that is released from BLOC-2–deficient endothelial cells has decreased binding to collagen and lacks HMW multimers, consistent with impaired biogenesis of WPBs. Impaired secretagogue-induced VWF release and multimerization of plasma VWF has been previously described specifically in BLOC-2−/− mice.19 Whether BLOC-2 also participates directly in WPB secretion as a component of the exocytic machinery remains uncertain. In striking contrast, exocyst clearly has distinct roles in WPB maturation vs WPB exocytosis. Exocyst, like BLOC-2, is required for production of mature, elongated WPB possessing HMW multimers. However, exocyst depletion augments WPB exocytosis, despite its role in WPB biogenesis. This observation indicates that exocyst separately regulates granule exocytosis in endothelium, acting as a clamp. Indeed, treatment of endothelial cells with Endosidin2, which interferes with EXO70, augmented VWF exocytosis within 2 hours, a period of time insufficient to impact WPB maturation.45 The presence of normal VWF multimers in Endosidin2-treated cells rules out any significant effects on WPB biogenesis. Furthermore, treatment of BLOC-2–deplete endothelial cells with Endosidin2 (Figure 7) or knockdown of EXOC4 (Figure 6) ‘rescued’ VWF exocytosis. These observations confirm that the exocyst complex has distinct roles in WPB maturation and exocytosis.
The exact molecular mechanism by which exocyst regulates exocytosis in endothelium is unknown, although three molecular mechanisms proposed to function in WPB exocytosis include proteins known to interact with exocyst. First, the Rab27a effector MyRIP is known to interact with exocyst components EXOC3 and EXOC4, linking exocyst complex to protein kinase A in mammalian cells.51 Endothelial MyRIP anchors maturing WPBs to the actin cytoskeleton via a tripartite complex with Rab27a and MyoVa, disruption of which augments VWF exocytosis.33,52 Rab27a, which also regulates WPB exocytosis more distally at the plasma membrane via its effector granulophilin, or Slp4-a, has also been shown to bind exocyst in a proteomics study, although the functional significance of this interaction has not been studied.49,53 Of note, although Slp4-a promotes VWF release in endothelial cells, it inhibits exocytosis in other cell types.49 Second, exocyst is also an established effector of small GTPase RalA.43,54 RalA, and its guanine exchange factor RalGDS, have been shown to be essential for both Ca2+ and cAMP-induced WPB exocytosis.55-57 Last, exocyst is known to modulate microtubule dynamics and negatively regulate tubulin polymerization in mammalian cells.42 The importance of microtubule on WPB trafficking and exocytosis has long been established.12,58,59 Although several proteins that facilitate WPB exocytosis bind to exocyst in mammalian cells, further studies will be required to determine how exocyst interacts to regulate WPB exocytosis.
Our data provide the first direct evidence of the role of the exocyst complex in endothelial WPB biogenesis and exocytosis. Figure 7F summarizes in a model our data that BLOC-2, in association with the exocyst complex, directs endosome-derived tubular transport carriers to maturing WPBs, thereby regulating endosomal transport essential for WPB biogenesis. At the level of VWF release, the exocyst complex independently controls WPB exocytosis by acting as a clamp. Release from the clamping activity of exocyst augments VWF release.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
For original data, please contact asharda@bidmc.harvard.edu.
Acknowledgments
The authors thank Michael S. Marks for reviewing this manuscript and providing thoughtful suggestions, Nikon Imaging Center at the Harvard Medical School for live cell imaging, and the Harvard Medical School EM facility for electron microscopy.
A.V.S. received a 2016 Mentored Research Award from the Hemostasis and Thrombosis Research Society, sponsored by Baxalta US Inc. R.F. received support from the National Institutes of Health, National Heart, Lung and Blood Institute (grants R01 HL125275 and R35 HL135775). J.E.I. received support from the National Institutes of Health, National Heart, Lung and Blood Institute (grants R01HL068130 and R01HL136394).
Authorship
Contribution: A.V.S. designed and performed experiments, analyzed data, and wrote the manuscript; A.M.B. performed experiments and edited the manuscript; J.A.H., A.R.W., C.F., L.M.M., and I.C.G. performed experiments and analyzed data; J.E.I. edited the manuscript; and R.F. designed experiments and edited the manuscript.
Conflict-of-interest disclosure: J.E.I. has financial interests in and is a founder of Platelet BioGenesis, a company that aims to produce donor-independent human platelets from human-induced pluripotent stem cells at scale. He is an inventor on this patent. The interests of J.E.I. were reviewed and are managed by the Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict-of-interest policies. R.F. has financial interests in and is a founder of PlateletDiagnostics. His interests are reviewed and managed by Beth Israel Deaconess Medical Center in accordance with their conflict-of-interest policies. The remaining authors declare no competing financial interests.
Correspondence: Robert Flaumenhaft, Division of Hemostasis and Thrombosis, Department of Medicine, Beth Israel Deaconess Medical Center, 330 Brookline Ave, Boston, MA 02215; e-mail: rflaumen@bidmc.harvard.edu.