Key Points
An SGLT2 inhibitor improves neutropenia/neutrophil dysfunction in GSD-Ib by lowering intracellular 1,5AG6P.
Empagliflozin, used in type 2 diabetes, is repurposed for neutrophil dysfunction in GSD-Ib without causing symptomatic hypoglycemia.

Abstract
Neutropenia and neutrophil dysfunction cause serious infections and inflammatory bowel disease in glycogen storage disease type Ib (GSD-Ib). Our discovery that accumulating 1,5-anhydroglucitol-6-phosphate (1,5AG6P) caused neutropenia in a glucose-6-phosphatase 3 (G6PC3)–deficient mouse model and in 2 rare diseases (GSD-Ib and G6PC3 deficiency) led us to repurpose the widely used antidiabetic drug empagliflozin, an inhibitor of the renal glucose cotransporter sodium glucose cotransporter 2 (SGLT2). Off-label use of empagliflozin in 4 GSD-Ib patients with incomplete response to granulocyte colony-stimulating factor (GCSF) treatment decreased serum 1,5AG and neutrophil 1,5AG6P levels within 1 month. Clinically, symptoms of frequent infections, mucosal lesions, and inflammatory bowel disease resolved, and no symptomatic hypoglycemia was observed. GCSF could be discontinued in 2 patients and tapered by 57% and 81%, respectively, in the other 2. The fluctuating neutrophil numbers in all patients were increased and stabilized. We further demonstrated improved neutrophil function: normal oxidative burst (in 3 of 3 patients tested), corrected protein glycosylation (2 of 2), and normal neutrophil chemotaxis (1 of 1), and bactericidal activity (1 of 1) under treatment. In summary, the glucose-lowering SGLT2 inhibitor empagliflozin, used for type 2 diabetes, was successfully repurposed for treating neutropenia and neutrophil dysfunction in the rare inherited metabolic disorder GSD-Ib without causing symptomatic hypoglycemia. We ascribe this to an improvement in neutrophil function resulting from the reduction of the intracellular concentration of 1,5AG6P.
Introduction
Glucose-6-phosphate translocase (G6PT/SLC37A4) and glucose-6-phosphatase 1 (G6PC1) are required for the conversion of G6P to glucose, ensuring glucose production by the liver and kidney.1 Unlike glucose-6-phosphatase, G6PT is expressed ubiquitously, and its deficiency2 underlies glycogen storage disease type Ib (GSD-Ib; MIM #232220), an autosomal, recessively inherited, rare (prevalence, ∼1 in 500 000)3 inborn error of carbohydrate metabolism.4 Patients present with hepatomegaly and severe hypoglycemia, for which the mainstay is strict dietary management.5 Neutrophils in GSD-Ib patients exhibit exaggerated apoptosis6 and have a dysfunctional metabolism, impairing protein glycosylation7 and resulting in neutrophil dysfunction,8 which leads to defective chemotaxis, oxidative burst, and bactericidal activity.9 Patients experience a high burden of neutrophil dysfunction–related symptoms, such as anourogenital lesions, infections, and inflammatory bowel disease (IBD).10 Subcutaneous injections of granulocyte colony-stimulating factor (GCSF) partially improve neutrophil numbers but not neutrophil dysfunction11 and involve a risk for monoclonal malignancies (myelodysplasia and acute myeloid leukemia).12,13
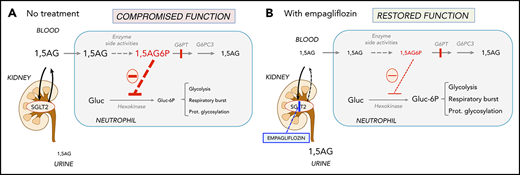
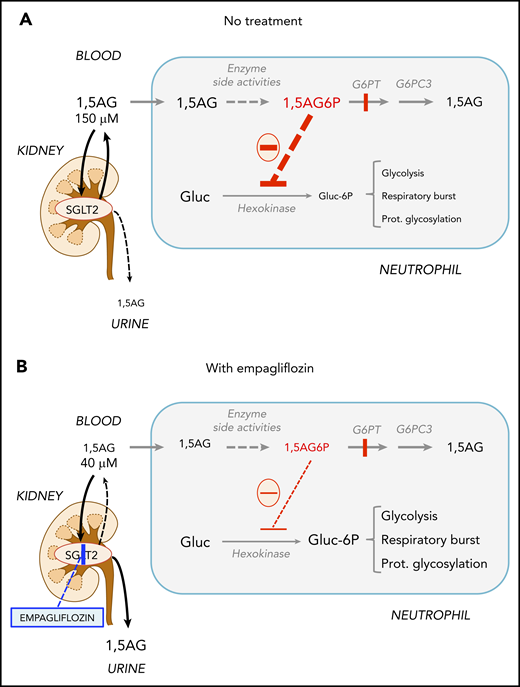
Recently, we found that G6PT transports not only G6P, but also its structural analog 1,5-anhydroglucitol-6-phosphate (1,5AG6P), which accumulates in the neutrophils of GSD-Ib patients (Figure 1). These neutrophils lack G6PT and therefore fail to transport 1,5AG6P from the cytosol into the endoplasmic reticulum, where it is normally dephosphorylated by G6PC3, a phosphatase in the membrane of the endoplasmic reticulum.14 Cytosolic accumulation of 1,5AG6P inhibits glucose phosphorylation by hexokinases and therefore glycolysis, the sole energy source for mature neutrophils.15,16 This mechanism explains neutrophil dysfunction and apoptosis not only in GSD-Ib, but also in G6PC3 deficiency,17 which underlies severe congenital neutropenia type 4 (MIM #612541).18
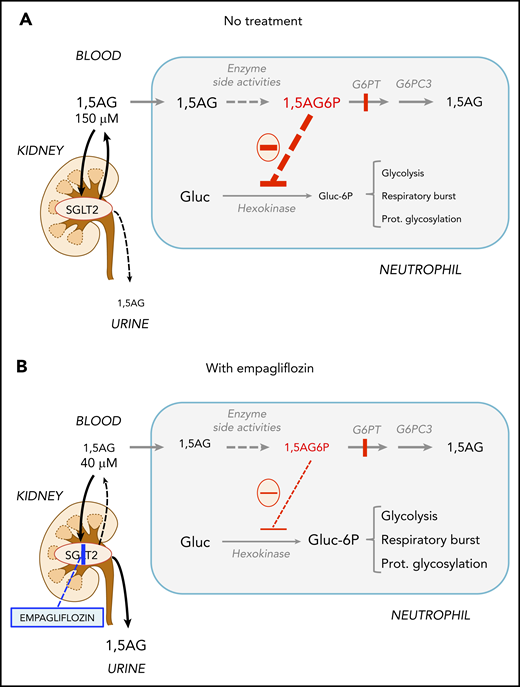
Empagliflozin lowers 1,5AG plasma levels and restores neutrophil function in GSD-Ib patients by lowering neutrophil 1,5AG6P. 1,5AG is a nondegradable glucose analog present in blood (∼150 µM). It is slowly phosphorylated to 1,5AG6P by the side activities of hexokinases and adenosine 5′-diphosphate–dependent glucokinase present in neutrophils. To prevent its accumulation, 1,5AG6P is transported into the endoplasmic reticulum by G6PT and dephosphorylated by the phosphatase G6PC3. (A) In GSD-Ib patients who are deficient in G6PT, 1,5AG6P accumulates in neutrophils. It is the rise in the concentration of 1,5AG6P that intoxicates neutrophils by strongly inhibiting hexokinases and depleting the intracellular pool of G6P (Gluc-6P) that is vital for neutrophils to survive and function.14 (B) Inhibiting the renal SGLT2 with empagliflozin leads to glucosuria by preventing the renal reabsorption of glucose, but also of 1,5AG, which results in its urinary excretion. Consequently, this leads to an approximate fourfold reduction in the concentration of 1,5AG in blood and of 1,5AG6P in neutrophils. This relieves the inhibition of hexokinases and increases the pool of G6P and of the metabolites in downstream pathways, improving glycolysis, respiratory burst, and protein glycosylation. Neutrophils function better, and neutropenia is partly corrected.
Empagliflozin lowers 1,5AG plasma levels and restores neutrophil function in GSD-Ib patients by lowering neutrophil 1,5AG6P. 1,5AG is a nondegradable glucose analog present in blood (∼150 µM). It is slowly phosphorylated to 1,5AG6P by the side activities of hexokinases and adenosine 5′-diphosphate–dependent glucokinase present in neutrophils. To prevent its accumulation, 1,5AG6P is transported into the endoplasmic reticulum by G6PT and dephosphorylated by the phosphatase G6PC3. (A) In GSD-Ib patients who are deficient in G6PT, 1,5AG6P accumulates in neutrophils. It is the rise in the concentration of 1,5AG6P that intoxicates neutrophils by strongly inhibiting hexokinases and depleting the intracellular pool of G6P (Gluc-6P) that is vital for neutrophils to survive and function.14 (B) Inhibiting the renal SGLT2 with empagliflozin leads to glucosuria by preventing the renal reabsorption of glucose, but also of 1,5AG, which results in its urinary excretion. Consequently, this leads to an approximate fourfold reduction in the concentration of 1,5AG in blood and of 1,5AG6P in neutrophils. This relieves the inhibition of hexokinases and increases the pool of G6P and of the metabolites in downstream pathways, improving glycolysis, respiratory burst, and protein glycosylation. Neutrophils function better, and neutropenia is partly corrected.
1,5AG6P is made by enzymatic side reactions from 1,5AG, a nondegradable glucose analog present in blood.19 Sodium glucose cotransporter 2 (SGLT2) inhibitors, such as empagliflozin, are antidiabetic drugs that inhibit renal glucose reabsorption in diabetic and normoglycemic patients20,21 and therefore cause urinary excretion of glucose. Glucosuria decreases renal 1,5AG reabsorption and lowers its serum concentrations.14,22-24
We recently showed that empagliflozin decreases intracellular 1,5AG6P in G6PC3-deficient mice and normalizes their absolute neutrophil counts (ANCs).14 Here we report the first results of repurposing empagliflozin in GSD-Ib patients to treat neutropenia and neutrophil dysfunction.
Patients and methods
Ethical considerations and study medication
This study evaluated the repurposing of empagliflozin as an innovative mechanism-based individualized treatment for 4 GSD-Ib patients. These patients were independently treated at 3 different tertiary hospitals and were experiencing severe clinical complications of neutrophil dysfunction despite standard treatment with GCSF. Informed oral and written consent to off-label treatment with empagliflozin was obtained from the patients or parents.
The SGLT2 inhibitor empagliflozin registered for type 2 diabetes in adults has a favorable safety profile. The most common adverse effects are increased thirst and urogenital fungal skin infections resulting from glucosuria. Urinary tract infection was reported more frequently in female patients treated with empagliflozin compared with placebo; there was no difference in male patients. This does not indicate per se a relevantly increased risk for infants or toddlers, but careful monitoring for signs of urinary tract infection in this population may be reasonable.
Hypoglycemia with empagliflozin has rarely been observed and only in coadministration with other antidiabetic drugs.25 In adults, pharmacokinetics and pharmacodynamics are well characterized, and a single daily dose of 10 to 25 mg is recommended. In obese adolescents, the same dosage was well tolerated, but published data as well as clinical experience are limited (more information in supplemental Methods, available on the Blood Web site). On the basis of these data and the biochemical rationale, we judged the risk/benefit ratio of empagliflozin use favorable in our patients.
Phenotypes of patients
The 4 patients had biallelic SLC37A4 variants, genetically confirming GSD-Ib (Table 1; Figure 2A). PT1, a 21-year-old woman, had constant oral and anourogenital lesions and infections. She had caries as a result of poor dental hygiene caused by painful aphthous gums. She had up to 20 bloody and watery stools daily, was almost exclusively tube fed, and complained of constant abdominal pain. She had microcytic anemia. Despite trials with various biologicals, hemicolectomy, and 10 µg/kg per day of GCSF, IBD and infections were not controlled. During the 5 months before empagliflozin treatment, median ANC was 0.5 × 109/L (reference range, 1.8 × 109/L to 8 × 109/L). Her adult Crohn disease activity score was 221, indicating severe disease.26
![Empagliflozin lowers plasma 1,5AG and intracellular 1,5AG6P and corrects neutropenia in GSD-Ib patients. (A) Time course of ANCs before and during empagliflozin treatment in 4 GSD-Ib patients. The top part of the right scale refers to the concentration of GCSF, and the bottom to empagliflozin. (B) Decrease in the concentration of plasma 1,5AG determined by liquid chromatography–mass spectrometry (LC-MS) during treatment. (C) Neutrophil 1,5AG6P determined in blood cells after centrifugation to remove plasma (for determination of 1,5AG) and normalized to total metabolite content (total ion current [TIC])14 and ANC. (D) 1,5AG6P in isolated granulocytes (polymorphonuclear neutrophils [PMNs]) and peripheral blood mononuclear cells (PBMCs) obtained from patient 1 (PT1) 1 day before and 28 or 50 days into empagliflozin treatment and compared with levels found in a healthy control (CT).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/9/10.1182_blood.2019004465/2/m_bloodbld2019004465f2.png?Expires=1768772884&Signature=dHG3CSYLrJSloIVzaD9B7Yei1xRT2z2RRuhfmpPs05dXtCESivXQB9zcQQqdrrCbg-eFpIak4l0UV9swboQ-If~aE-4LY9KCnYF4G2Fcd054w2PW4ym2XPX37kNFbRsZbw0yVMERvsKKmYx1WDtyL-AYUTuMBCbzhg2YExieJ5YHyTGqwsbrTa9i~zQaQGTgNZnNVjVwIG-JFJ9JIR-oLpcWLENJvxMW2eHQCXDfgQfKuXl4jfo2aCWijtfXZX5bvz6O14oQiD~L-NIfIvXYqjfac~WPpK3feDkRC3UItkAltqK3SPcLgp4mvQVqpkzTuovoS7TjJoP~jDJb3TAa9g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Empagliflozin lowers plasma 1,5AG and intracellular 1,5AG6P and corrects neutropenia in GSD-Ib patients. (A) Time course of ANCs before and during empagliflozin treatment in 4 GSD-Ib patients. The top part of the right scale refers to the concentration of GCSF, and the bottom to empagliflozin. (B) Decrease in the concentration of plasma 1,5AG determined by liquid chromatography–mass spectrometry (LC-MS) during treatment. (C) Neutrophil 1,5AG6P determined in blood cells after centrifugation to remove plasma (for determination of 1,5AG) and normalized to total metabolite content (total ion current [TIC])14 and ANC. (D) 1,5AG6P in isolated granulocytes (polymorphonuclear neutrophils [PMNs]) and peripheral blood mononuclear cells (PBMCs) obtained from patient 1 (PT1) 1 day before and 28 or 50 days into empagliflozin treatment and compared with levels found in a healthy control (CT).
Empagliflozin lowers plasma 1,5AG and intracellular 1,5AG6P and corrects neutropenia in GSD-Ib patients. (A) Time course of ANCs before and during empagliflozin treatment in 4 GSD-Ib patients. The top part of the right scale refers to the concentration of GCSF, and the bottom to empagliflozin. (B) Decrease in the concentration of plasma 1,5AG determined by liquid chromatography–mass spectrometry (LC-MS) during treatment. (C) Neutrophil 1,5AG6P determined in blood cells after centrifugation to remove plasma (for determination of 1,5AG) and normalized to total metabolite content (total ion current [TIC])14 and ANC. (D) 1,5AG6P in isolated granulocytes (polymorphonuclear neutrophils [PMNs]) and peripheral blood mononuclear cells (PBMCs) obtained from patient 1 (PT1) 1 day before and 28 or 50 days into empagliflozin treatment and compared with levels found in a healthy control (CT).
PT2, a 2-year-old girl, presented with constant oral and anourogenital lesions from birth, despite up to 10 µg/kg per day of GCSF from 3 months of age. She had 1 to 2 soft stools daily and was exclusively tube fed. Under GCSF, median ANC was 0.8 × 109/L (reference range, 1.8 × 109/L to 8 × 109/L). She had microcytic anemia, which was not responding to iron supplementation.
PT3, a 6-year-old boy, had presented at age 2 days with an infection complicated by convulsions as a result of infarction of the right medial and posterior cerebral arteries. Since then, he had been treated with antiepileptics in different combinations for focal seizures. He had had since birth frequent symptomatic hypoglycemias regularly leading to seizures, watery stools, multiple respiratory tract infections, and frequent ulcerative gingivitis. GCSF was started at age 16 months. Throughout his life, his median ANC was 1.8 × 109/L (reference range, 1.6 × 109/L to 7.6 × 109/L). At age 6 years, the clinical course was complicated by progressive painful abdominal distension and organomegaly, 5 to 9 watery stools per day, transfusion-dependent anemia, and severe hypoglycemias despite continuous gastric drip feeding, GCSF at up to 7 µg/kg per day, and 40 mg/kg of sulfasalazin twice daily. His pediatric Crohn disease activity score was 67.5 (severe disease).27
PT4, a 2-year-old girl, had recurrent mouth ulcers and abscesses in addition to persistent inflammation around the gastric tube side, which occasionally developed into erysipelas requiring antibiotic treatment. She had persistent diarrhea with up to 8 watery stools daily and recurrent vomiting multiple times per day, despite sulfasalazin treatment (2 mg/kg twice daily). She received up to 2.4 µg/kg per day of GCSF and had a median ANC of 0.5 × 109/L (reference range, 1.8 × 109/L to 5.4 × 109/L).
Detection and quantification of 1,5AG in plasma
1,5AG in plasma was quantified by LC-MS as previously described.14 Plasma was isolated after centrifugation (5 minutes at 500 g; 22°C) of 0.4 mL of freshly collected EDTA blood and kept at −80°C until analysis. Details can be found in supplemental Methods.
Detection and quantification of 1,5AG6P
To get an estimate of the 1,5AG6P concentration in granulocytes, this metabolite was quantified by LC-MS,14 as described in detail in supplemental Methods. The values for 1,5AG6P shown correspond to extracted-ion chromatograms of the [M-H]− form, and the areas under the curve (m/z = 243.0273) were normalized to (1) total ion current,14 which takes into account any variability in the sensitivity of the LC-MS measurements, and (2) the ANC present in the respective blood sample, because most of the 1,5AG6P is present in the neutrophils.14 The values obtained in this way are proportional to the intracellular concentration of 1,5AG6P in neutrophils.
Detection and quantification of glycosylated LAMP2 in isolated granulocytes (PMN) by western blot
Protein glycosylation was estimated in neutrophils by western blot analysis of LAMP2.28 For this, PMNs from PT1, PT3, and a healthy control were isolated from 5 mL of EDTA blood as described for quantification of 1,5AG6P.14 Preparation of protein extracts and analysis are detailed in supplemental Methods.
Neutrophil function assays of oxidative burst, chemotaxis, and bactericidal activity
Neutrophil function was assessed for (1) oxidative burst in 2 different laboratories using flow cytometric analysis, (2) chemotaxis, and (3) bactericidal activity as previously described.29-31 The detailed procedures for the analysis are available in supplemental Methods.
Results
Clinical course under empagliflozin
Patients’ clinical courses are summarized in Table 1. PT1 started with 5 mg of empagliflozin once daily (0.1 mg/kg per day) 48 hours after the last dose of GCSF. Glucose values in the first 10 days were measured by continuous glucose monitoring (CGM) and additional capillary measurements every 2 hours. In the following 30 days, capillary glucose was measured before every meal and later only upon clinical indication. Nine hours after the first empagliflozin dose, 1 asymptomatic mild hypoglycemia (2.7 mmol/L) occurred and was corrected with tube feeds. Thereafter, no hypoglycemia was observed (capillary values, 3.6-7.9 mmol/L) during the 288 days of follow-up. Empagliflozin was increased to 10 mg once daily (0.2 mg/kg per day) on day 2, and from day 7 onward, 2 daily doses of 10 mg were administered (0.4 mg/kg per day).
After 3 days of treatment, the patient reported increased appetite and ability to brush her teeth without pain or bleeding. After 5 days, a 4-week-old nonhealing genital wound, resulting from the opening of an abscess, finally closed. Since then, the patient had 5 self-limiting genital lesions, but no skin or mucosa lesions or infections. The stool frequency went down to 1 to 2 times daily within a week, and calprotectin levels in feces normalized. Her (adult) Crohn disease activity score decreased from 221 (day 0) to 138 (day 12) to 60 (day 288); values <150 are considered remission.26 Her weight increased by 5 kg, and her anemia resolved. At day 288, macrogol 3350 (13.7 g once daily) was started because of painful hard stools. The splenomegaly decreased from 17 to 15 cm in longitudinal diameter.
PT2 started empagliflozin at 5 mg once daily (0.4 mg/kg per day); GCSF was continued on days 2, 4, and 6 and was then discontinued by her parents because of clinical improvement. Capillary glucose was measured before every meal in the first 10 days and later only upon indication. All values were within the normal range during the 217 days of follow-up. The girl had increased appetite and started eating partly orally. She had no more skin or mucosa lesions. After 10 days, oral macrogol 3350 (6.9 g once daily) was started because of painful hard stools. Her anemia resolved.
PT3 started with 5 mg of empagliflozin once daily (0.2 mg/kg per day on days 1-8) and was increased to 10 mg (0.4 mg/kg per day on days 9-37) and 12.5 mg in 2 daily doses (0.5 mg/kg per day on days 38-83) while continuing GCSF at 5.3 μg/kg per day. From day 12, stools gradually became more solid and less frequent, and at day 15, continuous gastric drip feeding was replaced by frequent meals with small doses of uncooked cornstarch. As determined by CGM, hypoglycemic episodes quickly became less frequent. Daily glucose urinary excretion on empagliflozin at 0.4 and 0.5 mg/kg per day corresponded to a weight percentage of 4% and 7%, respectively, of the daily dose of uncooked cornstarch.
From day 29, GCSF was gradually tapered, and the boy became more active and started to eat by himself for the first time in his life; the nasogastric tube could be removed, and fasting intervals of ∼3 hours were tolerated. Abdominal circumference and organomegaly decreased, and no more blood transfusions were needed.
During the additional follow-up until day 246, he had several episodes of intercurrent infections (eg, day 106 otitis media requiring amoxicillin, day 168 upper respiratory infection), which led to a worsening of the general clinical situation, with reoccurrence of oral aphthous lesions and loose stools. He had not shown any symptomatic hypoglycemias, including hypoglycemia-induced seizures, since the start of empagliflozin. Several attempts to taper and stop GCSF were made, but GCSF had to be reintroduced during periods of infection. With sulfasalazine in unchanged dosage (80 mg/kg per day), empagliflozin at a final dosage of 0.7 mg/kg per day taken in 2 10-mg doses and 1.4 μg/kg of GCSF daily, the patient was currently in a stable clinical situation. He had no more abdominal pain, ate mainly by himself, and had 2 to 3 formed stools daily. The hemoglobin levels nearly normalized, no more transfusions were needed, and splenomegaly decreased from 15.1 to 12.3 cm in longitudinal diameter. His pediatric Crohn disease activity score decreased from 67.5 (severe disease) to 17.5 (day 61), 22.5 (day 120), and 20 (day 246; mild disease).27
PT4 started with empagliflozin at 5 mg per day (0.3 mg/kg per day), while GCSF was continued unchanged at 2.4 μg/kg per day. On CGM, during the 191 days of follow-up, she showed occasional low glucose levels (2.8-3.9 mmol/L); none were symptomatic, and they occurred at a lower frequency than before treatment. She had no more skin or mucosa lesions. During treatment, she had 2 well-tolerated viral infections and 1 otitis media incident treated with amoxicillin. Her stooling frequency improved to twice daily, and stools were better formed. Empagliflozin was increased to 7.5 mg once daily (0.5 mg/kg per day) on day 98, sulfasalazine was stopped (day 142), and GCSF (day 133) was decreased to 3 times weekly at 2.4 μg/kg per dose instead of daily use. Her hemoglobin level improved significantly, and C-reactive protein level normalized.
Neutrophils, plasma 1,5AG, intracellular 1,5AG6P, and neutrophil function
Before treatment with empagliflozin, all 4 patients showed wide fluctuations in ANC, which sometimes fell to very low values (Figure 2A). In PT1, the marked drop in ANC after interruption of GCSF and the start of empagliflozin was followed by a slow increase of ANC to 0.5 × 109/L to 0.9 × 109/L. In PT2, in whom GCSF was interrupted 6 days after starting empagliflozin, ANC value eventually normalized; a transient but fully reversible drop occurred when empagliflozin was transiently reduced from 5 mg (0.4 mg/kg per day) to the same dose 3 times weekly, indicating a tight dose-effect relation in the chosen dose range. In PT3, ANC remained stable despite gradually tapering GCSF from 51.1 to 9.8 µg/kg per week (−81%). In PT4, median ANC increased from 0.5 × 109/L to 1.1 × 109/L before tapering of GCSF treatment from 16.8 to 7.2 µg/kg per week (−57%). Overall, treatment with empagliflozin tended to decrease the amplitude of the fluctuations in ANC, with the lowest ANC during empagliflozin treatment being generally higher than the lowest ANC observed on GCSF before empagliflozin treatment.
Daily empagliflozin intake resulted in a four- to fivefold decrease in plasma 1,5AG in all patients and a new stable concentration of 1,5AG (34 to 45 µM for PT1 to PT3) after 2 to 3 weeks (Figure 2B). Levels in PT4 decreased from 301 to <90 µM within a month. To estimate the 1,5AG6P accumulation in neutrophils, we determined its amount in whole blood and normalized this value to the ANCs. In PT1 and PT2, intracellular 1,5AG6P decreased by ∼75% in parallel (Figure 2C) with plasma 1,5AG (Figure 2B). This decrease in 1,5AG6P was less apparent in PT3. Of note, for PT3, we compared 1,5AG6P in blood (Figure 2C) with that in isolated leukocytes. 1,5AG6P in PT3 leukocytes showed a fourfold reduction (supplemental Figure 1) similar to that seen for PT1 and PT2 in whole blood (Figure 2C).
This reduction of 1,5AG6P in leukocytes was further confirmed in an isolated preparation of granulocytes (PMNs) and lymphocytes (peripheral blood mononuclear cells) for PT1. As we had previously observed,14 1,5AG6P was approximately fourfold more abundant in PMNs than in peripheral blood mononuclear cells. Furthermore, on day 28, the treatment lowered 1,5AG6P in PMNs by approximately fourfold, consistent with a similar decrease in plasma 1,5AG. Interestingly, the amount of 1,5AG6P in PMNs of PT1 still remained ∼25-fold above that of a healthy control (Figure 2D), suggesting that the metabolic changes in the neutrophils were only partly corrected (Figure 3A). This finding is consistent with the observation that glycosylation of LAMP2 in PMNs from both PT1 and PT3 (Figure 3B) was remarkably improved during treatment, but not fully normalized.
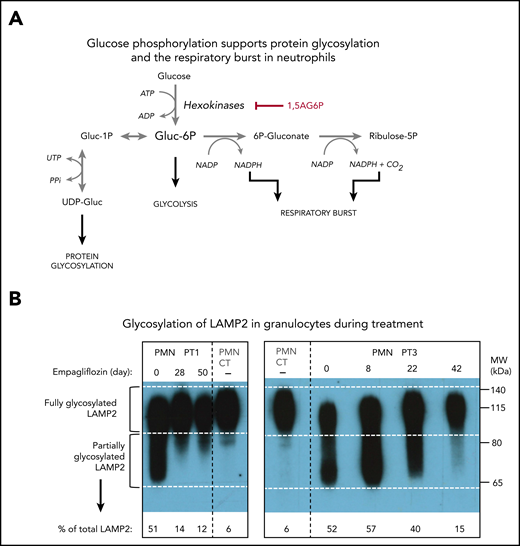

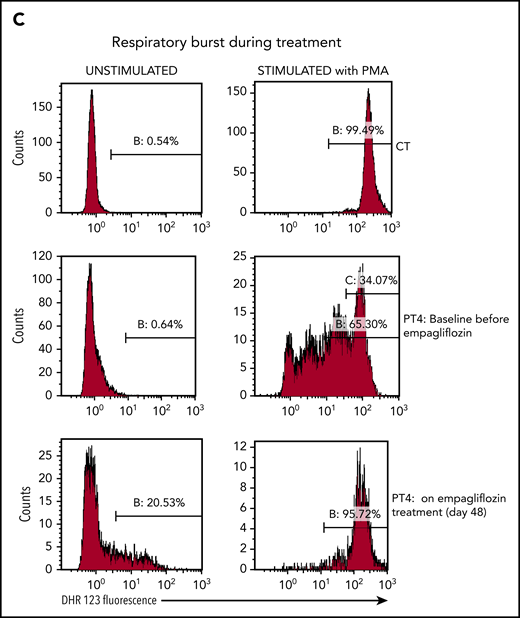
Empagliflozin corrects protein glycosylation and oxidative burst in GSD-Ib patients after empagliflozin treatment. (A) Role for G6P (Gluc-6P) in uridine diphosphate glucose (UDP-Gluc) production (essential for protein glycosylation) and reduced NAD phosphate (NADPH) production (essential for the respiratory burst reactions). (B) Western blots illustrating the almost complete correction of glycosylation for the protein LAMP2 in granulocytes (PMNs) from PT1 and PT3 isolated before and during empagliflozin treatment and compared with a healthy control (CT). (C) Empagliflozin treatment corrected the defective respiratory burst in PT4. Whole blood was stimulated with phorbol myristate acetate (PMA) at 37°C for 15 minutes. Red blood cells were lysed, and the sample was analyzed immediately by flow cytometry with gating set on neutrophils. Gate B: neutrophils showing increased DHR123 fluorescence compared with background fluorescence in unstimulated cells. Gate C: PT4 PMA-stimulated neutrophils showing DHR123 fluorescence equivalent to that of the date-matched normal control. ADP, adenosine 5′-diphosphate; ATP, adenosine triphosphate; MW, molecular weight; PPi, inorganic pyrophosphate; UTP, G1P uridylyltransferase.
Empagliflozin corrects protein glycosylation and oxidative burst in GSD-Ib patients after empagliflozin treatment. (A) Role for G6P (Gluc-6P) in uridine diphosphate glucose (UDP-Gluc) production (essential for protein glycosylation) and reduced NAD phosphate (NADPH) production (essential for the respiratory burst reactions). (B) Western blots illustrating the almost complete correction of glycosylation for the protein LAMP2 in granulocytes (PMNs) from PT1 and PT3 isolated before and during empagliflozin treatment and compared with a healthy control (CT). (C) Empagliflozin treatment corrected the defective respiratory burst in PT4. Whole blood was stimulated with phorbol myristate acetate (PMA) at 37°C for 15 minutes. Red blood cells were lysed, and the sample was analyzed immediately by flow cytometry with gating set on neutrophils. Gate B: neutrophils showing increased DHR123 fluorescence compared with background fluorescence in unstimulated cells. Gate C: PT4 PMA-stimulated neutrophils showing DHR123 fluorescence equivalent to that of the date-matched normal control. ADP, adenosine 5′-diphosphate; ATP, adenosine triphosphate; MW, molecular weight; PPi, inorganic pyrophosphate; UTP, G1P uridylyltransferase.
In PT4, the oxidative burst index before treatment was 143, with only 34% neutrophils having an oxidative burst response equivalent to that of the healthy control sample. It improved after 48 days of treatment to 178, with 96% of neutrophils showing a good oxidative burst (Figure 3C). On empagliflozin, neutrophil chemotaxis showed a mean migration, with a buffer of 33 μm (reference range, 24-54 µm) and with activated serum of 69 μm (reference range, 68-114 µm), and a bactericidal assay showed a normal killing of 86% (reference, >71%). Likewise, when PT1 and PT2 were treated with empagliflozin, their PMNs responded to phorbol myristate acetate almost as well as the corresponding date-matched control samples (response in 89.7% and 94.5% of patients’ PMNs vs 96.8% and 98.8% in date-matched controls; supplemental Figure 2A-B). However, when the respiratory burst of an untreated GSD-Ib patient was tested in the same conditions, only 77.8% of the patient’s PMNs responded to the phorbol myristate acetate stimulation, compared with 96.9% of those from a date-matched control (supplemental Figure 2C). This suggests that with empagliflozin, the function of neutrophils from PT1 and PT2 was also largely normalized.
Discussion
We describe a novel mechanism-based treatment of neutropenia and neutrophil dysfunction in 4 patients with GSD-Ib. This treatment is based on the SGLT2 inhibitor empagliflozin decreasing the concentration of 1,5AG in plasma and thereby reducing the concentration of its toxic derivative 1,5AG6P in neutrophils. Empagliflozin dramatically improved the heterogeneous clinical findings related to neutrophil dysfunction in all 4 patients. Furthermore, it allowed the termination of GCSF in 2 patients and reduction by 57% to 81% in the 2 others without decrease in ANC.
Treatment with empagliflozin ended the abdominal pain and frequent watery stools that seriously affected patients’ daily life activities and allowed healing of the oral and anourogenital lesions and infections. Even though the scoring systems designed for Crohn colitis have their limitations in GSD-Ib patients, the trend of score improvement was unquestionable in the 2 patients with overt IBD. Furthermore, hemoglobin levels improved in all patients, anemia resolved in 3 of 3 patients, and 3 of 4 patients were (partly) weaned from tube feeding.
A surprising observation of the present study is that the clinical improvement observed during empagliflozin treatment was not accompanied by an obvious increase in ANC compared with the situation where the patients were treated with GCSF alone. This was strikingly the case for PT1, in whom a clinical improvement had already been seen while her ANC value was still <0.2 × 109/L. These findings suggest that the improvement of the clinical state was largely due to a recovery in the functions of neutrophils. Accordingly, we found that the abnormal glycosylation7,32 in the neutrophils of the 2 GSD-Ib patients tested (PT1 and PT3) was largely (although not completely) corrected during empagliflozin treatment (Figure 3B). In PT4, we documented normalization of the oxidative burst defect unresolved by GCSF treatment, and in PT1 and PT2, we observed a normal oxidative burst under treatment11 (Figure 3C; supplemental Figure 2). Likewise, on empagliflozin treatment, neutrophils from PT4 showed normal chemotaxis and bactericidal activity, which are deficient in a vast majority of GSD-Ib patients.9 Of note, neutrophils obtained from GCSF-treated patients were previously found to be dysfunctional11 and to show a marked glycosylation defect.7 Similarly, neutrophils from GSD-Ib mice treated with GCSF continued to exhibit impaired cell adhesion and migration.33 This difference is understandable, because GCSF treatment, unlike empagliflozin treatment, is not expected to reduce the accumulation of the metabolic inhibitor 1,5AG6P but only improve overall neutrophil function by increasing the number of (dysfunctional) neutrophils. Improvement of the neutrophil function is likely to help recruit neutrophils at the sites of infection and increase their capacity to kill bacteria, thus explaining at least part of the impressive clinical benefit observed in the patients.
In addition, Figure 2D shows that it is likely that lowering blood 1,5AG could equally reduce 1,5AG6P in other peripheral blood mononuclear cells like macrophages or T cells that also seem to be involved in immune cell homeostasis in GSD-Ib patients.3,34 Therefore, it is possible that empagliflozin treatment has benefits that go beyond the improvements in neutrophil protein glycosylation7,32 and function that we have documented here.
As judged from PT1 and PT2, in whom GCSF treatment was replaced by empagliflozin, both treatments have roughly the same beneficial effect on ANC (Figure 2A), except that the fluctuations in ANC observed with GCSF treatment are attenuated on empagliflozin. In PT4, on an unchanged GCSF dose, there was a significant increase in ANC. Furthermore, a positive effect of empagliflozin on ANC values may also be deduced from the data shown for PT3 and PT4, because empagliflozin treatment allowed for a decrease in GCSF dosage without reduction in ANC. The neutropenia found in G6PT deficiency is due both to a decrease in production of mature neutrophils and to an increase in their apoptosis.6,8,35 Both are most likely the consequence of the decrease in the rate of glucose metabolism14 in cells that are highly dependent on glucose.15,16 By reducing the intracellular concentration of the hexokinase inhibitor 1,5AG6P, empagliflozin is expected to improve the production of mature neutrophils and reduce their apoptosis rate. We hypothesize that the reduction in the amplitude of ANC fluctuation with empagliflozin treatment may result from an increase in the lifetime of neutrophils.
Empagliflozin was well tolerated up to 0.7 mg/kg per day and did not induce significant, symptomatic hypoglycemias within the observational period of up to 288 days (942 patient-days in total). On the contrary, empagliflozin improved glucose homeostasis in PT3, who did not show any more hypoglycemia-induced seizures, and PT4. This suggests that IBD could play an important role in poor glycemic control in GSD-Ib, possibly by limiting intestinal glucose absorption. We prioritized the prevention of hypoglycemia by a careful gradual dose increase of empagliflozin over a progressive dose-finding scheme. Nevertheless, a clear dose response was observed in PT2. Thus, the selected final doses of 0.3 to 0.7 mg/kg per day in 1 or 2 daily doses seem to be within the therapeutic window. For now, it remains unclear if higher doses could be tolerated or more effective.
Another advantage of empagliflozin treatment is that painful daily GCSF injections were no longer necessary in 2 patients and were significantly reduced in the other 2 patients. Minimizing GCSF administration reduces both the increased risk for malignancies under long-term GCSF treatment12 and cost, because empagliflozin is at least 10-fold less expensive than GCSF treatment.
The presented data indicate that empagliflozin seems promising as treatment for neutrophil dysfunction in children and adults with GSD-Ib. It could also be useful in treating the severe neutropenia and neutrophil dysfunction present in G6PC3 deficiency,7,18,36 which equally originates from the accumulation of 1,5AG6P in neutrophils.14 This is a serious condition, for which allogeneic stem cell transplantation has recently been tested.37 Of note, we have previously shown that empaglifozin treatment in G6PC3-deficient mice normalized neutrophil counts.14 We have not addressed the problem of neutrophil dysfunction, but because neutropenia proceeds from a mechanism similar to that in GSD-Ib, it is likely that this dysfunction would also be corrected.
Future work should aim at better understanding the origin of 1,5AG and determining whether it is feasible to decrease its production. Classical knowledge is that ∼90% of this polyol is food derived38 ; however, the finding that the 1,5AG level is linked to genomic loci encoding intestinal glycosidases39 suggests that the endogenous origin, possibly in the intestine, may have been underestimated.
The individual off-label empagliflozin treatment of 4 GSD-Ib patients provides the preliminary data to develop an international multicenter (randomized placebo-controlled) clinical trial to evaluate and monitor safety, efficacy, and patient outcomes in a larger cohort. Such information is warranted, because there is considerable heterogeneity among GSD-Ib patients, as already shown the 4 patients presented here. Additional studies must also focus on the safety of the use of SGLT2 inhibitors in children in general, because no data are available yet for this age group, particularly regarding long-term use. It is encouraging that the treatment occurred without noted adverse effects in 2 patients age 2 years as well as in a young adult. Clinicians need to be particularly aware of possible hypoglycemic episodes and should observe kidney function, because changes in kidney function have been described as a long-term complication in GSD-Ib.
Treating neutropenia, infections, complications, and adverse effects is proposed as a main research priority for GSD-Ib patients.40 Since presenting our data at the 5th International Glycogen Storage Disease Conference in 2019 (Brazil), we have been contacted by numerous physicians with experience in GSD-Ib who are also planning to treat their patients. An international multicenter (randomized placebo-controlled) trial will obviously be the best way to thoroughly evaluate this new therapy, which may potentially present risks that are not yet appreciated. However, given the known challenges for trials in rare diseases41 and the urgent need for treatment in severely affected patients, alternative trial designs (eg, n = 1 trials) should be taken into account. Additionally, we have initiated a patient registry to systematically capture the safety and efficacy of this new treatment approach in children and adults with GSD-Ib when they are treated on an individual basis outside a clinical trial.
Our work illustrates the importance of metabolite repair, a new and quickly expanding concept in intermediary metabolism; metabolite repair enzymes destroy the toxic side products resulting from lack of enzyme specificity. Seven inborn errors of metabolite repair have now been identified.17,42 The neutropenias of GSD-Ib and G6PC3 deficiency are the first that seem to be treatable.
In summary, treating a rare disease known to cause hypoglycemia with a glucose-lowering drug used in type 2 diabetes seems counterintuitive. However, the goal of the treatment is to reduce a glucose analog that is toxic to neutrophils in GSD-Ib patients. Thus, this study underlines the importance of a thorough understanding of human intermediary metabolism, which has allowed a rapid and successful clinical translation through the repurposing of an existing drug.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Karol Kerr (Center for Cancer and Blood Disorders, Children’s Hospital Colorado Springs, Colorado Springs, CO), Peter Baker II (Inherited Metabolic Diseases Clinic, Children’s Hospital Colorado, Aurora, CO), and Johannes Spenger (University Children’s Hospital, Paracelsus Medical University, Salzburg, Austria) for their shared care of the patients. The authors also thank Anne Roscher (University Children’s Hospital, Paracelsus Medical University, Salzburg, Austria) and Dorothea Möslinger and Vassiliki Konstantopoulou (University Children’s Hospital Vienna) for fruitful discussions.
This work was supported by Walloon Excellence in Lifesciences and Biotechnology (WELBIO) (CR-2015A-09) and the European Union’s Horizon 2020 Research and Innovation Program under the European Research Area Network (ERA-NET) Cofund action #643578 (E.V.S.), and by the Fonds National de la Recherche Scientifique (FNRS; J.0104.18) (M.V.-d.-C.). M.V.-d.-C. is a chercheur qualifié (qualified researcher) of the FNRS. This work was also supported by the E-Rare project GENOMIT (Austrian Science Fund FWF, I 2741-B26) (J.A.M.) and by the Anniversary Fund of the Oesterreichische Nationalbank (OeNB, 18023) (S.B.W.).
Authorship
Contribution: S.B.W., E.V.S., and M.V.-d.-C. conceived the study and wrote the manuscript; S.B.W., J.L.K.V.H., T.G.J.D., E.O., J.A.M., F.J.v.S., F.B.L., and S.G. acquired and analyzed clinical data; M.V.-d.-C. and N.C. acquired and analyzed all biochemical data on 1,5-anhydroglucitol, 1,5-anhydroglucitol-6-phosphate, and protein glycosylation; V.K. and A.K. performed the functional assays on neutrophils; and all authors had full access to all data, reviewed drafts of the manuscript, and approved the final version.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Saskia B. Wortmann, University Children’s Hospital, Paracelsus Medical University, Mullner Hauptstrasse 48, 5020 Salzburg, Austria; e-mail: s.wortmann@salk.at; and Maria Veiga-da-Cunha, de Duve Institute, UCLouvain, 75 Av. Hippocrate, B-1200 Brussels, Belgium; e-mail: maria.veiga@uclouvain.be.

![Empagliflozin lowers plasma 1,5AG and intracellular 1,5AG6P and corrects neutropenia in GSD-Ib patients. (A) Time course of ANCs before and during empagliflozin treatment in 4 GSD-Ib patients. The top part of the right scale refers to the concentration of GCSF, and the bottom to empagliflozin. (B) Decrease in the concentration of plasma 1,5AG determined by liquid chromatography–mass spectrometry (LC-MS) during treatment. (C) Neutrophil 1,5AG6P determined in blood cells after centrifugation to remove plasma (for determination of 1,5AG) and normalized to total metabolite content (total ion current [TIC])14 and ANC. (D) 1,5AG6P in isolated granulocytes (polymorphonuclear neutrophils [PMNs]) and peripheral blood mononuclear cells (PBMCs) obtained from patient 1 (PT1) 1 day before and 28 or 50 days into empagliflozin treatment and compared with levels found in a healthy control (CT).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/9/10.1182_blood.2019004465/2/m_bloodbld2019004465f2.png?Expires=1768779655&Signature=1tc4DheKXpnbv3vqCqemG13FbP9F7Z7QH-RIuY3Yrlpg4NYyZCIXenIjNA-UtCn8fYnmW5w5VWbN6VuBjo7AQH6xa-ojfe2EQyxDb6IloHd4dCzGkmgqCxcRQQMceizc8Mxd~4-w9SJJnFwvZjEKhZPnKv7RFJr0BKhtsOwcdluzYIqeHhniLASMPwRRHHVTDaVrUlRpJO9HKl9C6F7EbMwfvW1jwxsH0l9x~mqSTCS3xts7I6QMsP25mggUF~~2uHNZ1cEZVASDP2jb8OP66XAyPHHCOGLvNJyXYbqkgXqhbZI85v4GqkykB241CIsvYhFGD9oO4um9tCO~-G8org__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)