In this issue of Blood, elegantly demonstrate that the proresolving protein annexin A1 (AnxA1) effectively dampens a proinflammatory neutrophil phenotype and, as a result, its important contribution to cerebral thromboinflammation in sickle cell disease (SCD).1
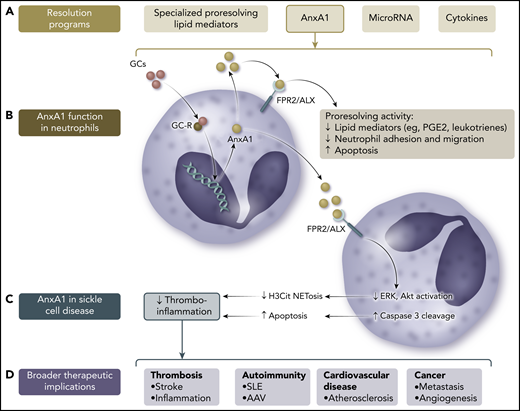
Role of AnxA1 in the resolution of neutrophil-dependent thromboinflammation in SCD and beyond. (A) Proresolving factors comprise lipid mediators, nucleic acids, and proteins, including cytokines and AnxA1. (B) AnxA1 is a glucocorticoid (GC)-regulated protein that mediates some of the anti-inflammatory effects of endogenous and synthetic GCs via its main receptor, FPR2/ALX. (C) In SCD, AnxA1 may act in neutrophils to downregulate the activity of ERK and Akt signaling pathways and promote cleavage of caspase 3, resulting in a switch from a proneutrophil extracellular trap (NET)-otic to apoptotic phenotype, thus reducing thromboinflammation. (D) Neutrophil reprogramming via AnxA1 and its related peptides may have therapeutic potential in a broad range of diseases where NETosis contributes to pathogenesis.
Role of AnxA1 in the resolution of neutrophil-dependent thromboinflammation in SCD and beyond. (A) Proresolving factors comprise lipid mediators, nucleic acids, and proteins, including cytokines and AnxA1. (B) AnxA1 is a glucocorticoid (GC)-regulated protein that mediates some of the anti-inflammatory effects of endogenous and synthetic GCs via its main receptor, FPR2/ALX. (C) In SCD, AnxA1 may act in neutrophils to downregulate the activity of ERK and Akt signaling pathways and promote cleavage of caspase 3, resulting in a switch from a proneutrophil extracellular trap (NET)-otic to apoptotic phenotype, thus reducing thromboinflammation. (D) Neutrophil reprogramming via AnxA1 and its related peptides may have therapeutic potential in a broad range of diseases where NETosis contributes to pathogenesis.
The last decade has seen an increasing recognition of resolution biology, the concept that inflammation resolution is an active and regulated process, which when ineffective can lead to bystander tissue injury during acute inflammation or allow progression to chronic inflammation and organ damage. There is now an appreciation of a broad repertoire of resolving programs, including cytokines (eg, transforming growth factor-β and interleukin-10), specialized proresolving lipid mediators (eg, lipoxins, maresins, protectins, and resolvins), some microRNAs, and AnxA1.
AnxA1 is a 37-kDa phospholipid-binding protein that is widely expressed in leukocytes and epithelial and endothelial cells. It is present both intracellularly and at the cell membrane, although its main described biological function is as a secreted protein that mediates some of the anti-inflammatory effects of endogenous and synthetic steroids.2 AnxA1 signals via the G protein–coupled formyl peptide receptor 2 (FPR2/ALX), which also serves as a receptor for lipoxin A4, another proresolving mediator. This receptor is predominantly expressed on leukocytes, where the proresolving effects of AnxA1 (and peptides derived from its N-terminal domain) include inhibiting the release of proinflammatory lipid mediators, modulating pro- and anti-inflammatory cytokine production, reducing neutrophil migration and accumulation, and promoting neutrophil clearance via apoptosis (through caspase 3) and efferocytosis.2 Altered circulating and tissue concentrations of AnxA1 have been described in a broad range of inflammatory diseases, including atherosclerosis, diabetes mellitus, inflammatory bowel disease, preeclampsia, and reperfusion injury, and AnxA1 has emerged as a target for therapeutic manipulation (see figure).
In the current work, Ansari et al investigate the role of the AnxA1/FPR2/ALX axis in thrombosis in SCD, using both a murine model and patient-derived samples. They show that provoked cerebral thrombosis in sickle transgenic mice (STM) is dependent upon circulating neutrophils and that administration of an N-terminal–derived AnxA1 mimetic peptide (Ac2-26) can attenuate thrombosis. Treatment with Ac2-26 resulted in a fall in circulating markers of NETosis in STM, and DNase treatment, which degrades NETs, had a similar therapeutic effect. In vitro, Ac2-26 treatment reduced markers of NET formation in murine SCD neutrophils. This effect was not seen in control neutrophils, suggesting a specific role of AnxA1 in resolving the proinflammatory phenotype acquired by neutrophils in the context of SCD. In translational studies, the authors found that patients with SCD had decreased circulating concentrations of endogenous AnxA1 and that, similar to murine SCD neutrophils, patient-derived neutrophils demonstrated enhanced markers of NETosis. This could be rescued in vitro by Ac2-26, without significant effects on healthy donor neutrophil responses. The authors go on to show that signaling from the FPR2/ALX receptor is dependent upon the ERK and Akt kinases, recognized as important mediators of NET release, and that Ac2-26 treatment upregulates cleaved caspase 3, suggesting a switch from a NETotic to an apoptotic response. Overall, these data support therapeutic targeting of the AnxA1/FPR2/ALX axis for prevention and treatment of thromboinflammation in SCD.
SCD provides a disease paradigm wherein neutrophils acquire a state of extreme activation. The current findings build on existing data suggesting these proinflammatory neutrophils, potentially via NETotic response to free heme,3 play a role in the recurrent episodes of microcirculatory vasoocclusion and ischemia reperfusion injury that contribute to organ damage in SCD.4 These data, therefore, may have broad implications, because the ability to reprogram neutrophils might have therapeutic potential in a range of diseases in which NETs are pathogenic. These include ischemic stroke and the thrombotic complications of many inflammatory diseases, in which NETs provide a stimulus and scaffold for clot formation, and autoimmune conditions, such as systemic lupus erythematosus (SLE)5 and antineutrophil cytoplasm antibody-associated vasculitis,6 where the aberrant extracellular expression of nuclear and cytoplasmic components contained in NETs are a source of potential autoantigen and immunostimulatory proteins. Broader still, NETs are implicated in the progression of both atherosclerosis, in which they polarize macrophages to a proinflammatory phenotype within lipid-rich plaque,7 and cancer, in which they are implicated in angiogenesis and metastasis.8
However, not all NETs are equal. They were first identified as part of the innate host defense response to infection. The mechanisms for NET release, and their constituents, differ between health and disease. In SLE, for example, NETs are spontaneously released and enriched for oxidized mitochondrial DNA, which may account for their greater immunogenicity.9 Notably, in the current study, Ac2-26 did not affect measures of healthy neutrophil NETosis, consistent with its proresolution rather than anti-inflammatory action. Alongside its other proresolving functions, AnxA1 makes for an appealing therapy that can modulate inflammation (and thrombosis) without undesirable immunosuppressive effects. Recent advances in the delivery of peptide therapies through the use of nanomedicines,10 which allow controlled spatiotemporal release in a site-specific manner, may enhance our ability to target therapy and avoid systemic effects even further.
Conflict-of-interest disclosure: The authors declare no competing financial interests.