

In this issue of Blood, describe their findings in a murine model of sickle cell anemia (SCA) in which mice were exposed to an iron (Fe)-restricted diet.1 This intervention resulted in improved hematocrit, with an increased number of red blood cells (RBCs) and a decreased mean corpuscular hemoglobin concentration (MCHC) and serum bilirubin. The tendency of the cells to sickle in vitro was reduced, and an improvement was also detected in the serum concentration of VCAM-1, a biomarker of endothelial activation.
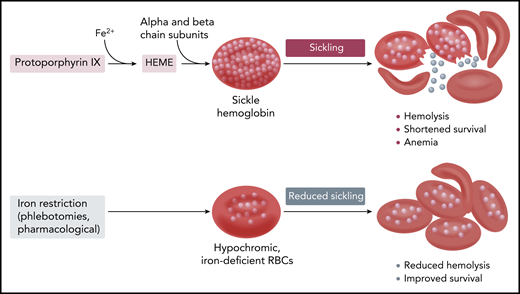
Hb synthesis and sickling in sickle erythrocytes with normal and reduced Hb content and concentration.
Hb synthesis and sickling in sickle erythrocytes with normal and reduced Hb content and concentration.
Although these effects were mild and did not significantly affect total hemoglobin (Hb) and reticulocyte count, this study in a well-established mouse model of the disease provides further support to the notion that, within certain limits, iron restriction may be beneficial for patients with sickle cell disease (SCD). The “iron hypothesis” was first put forward by Lincoln et al in 19732 and finds its pathophysiological basis on the kinetics of Hb polymerization and its unique dependence on Hb concentration. Hypochromic cells have a reduced Hb concentration, and thus in SCD, their Hb polymerization is delayed and they are less likely to sickle. It has also been known for a long time that coinheritance of diseases that ultimately reduce both Hb content (MCH) and concentration (MCHC) is a powerful modifier of SCD (see figure): Hb S/β thalassemia is recognized as a generally less-severe (but not complication-free) form of sickle cell anemia, whereas homozygous sickle cell anemia with associated α thalassemia. Hb S/β thalassemia has been known to exhibit less hemolysis, and possibly because of the higher Hb values, more frequent vaso-occlusive complications such as bone disease and painful crises.
The iron-restriction hypothesis was championed for many years by Oswaldo Castro at Howard University and has found many followers in France, especially for the treatment of patients heterozygous for both Hb S and Hb C (Hb SC disease). Hb SC disease is characterized by a milder clinical phenotype, but it still has significant complications related to higher cellular Hb concentration, higher Hb values (>10 g/dL in most patients) resulting in increased blood viscosity, and symptoms related to vaso-occlusion (painful crises, retinopathy, sensorineural otological disease, avascular necrosis, and nephropathy).3
In several case reports of patients affected by SCA, Castro et al showed that iron deficiency decreased hemolysis (lower serum bilirubin and lactate dehydrogenase and lower reticulocyte counts), reduced the fraction of dense cells, and improved RBC survival.4-6 Clinical improvement was also described with iron deficiency and worsening of symptoms with iron replacement therapy. Rombos et al7 showed similar hematologic and clinical improvements in 13 Greek patients, most of them affected by Hb S/β thalassemia. In a large retrospective study in patients with Hb SC disease, Lionnet et al8 showed measurable, but uncontrolled, clinical improvements in 71% of patients treated with regular phlebotomies. More recent studies have also highlighted a connection between nocturnal hypoxia and higher iron availability (estimated from transferrin saturation), again suggesting the deleterious effect of iron on SCD, although the investigators of this study did not invoke the mechanisms described above.9
The iron hypothesis has been in existence for at least 4 decades, but no controlled, properly designed studies have been carrier out to test it. Patients affected by SCD have an iron metabolism poised toward increased absorption, which ultimately leads to iron overload. Thus, an iron-deficient state can be induced only with repeated phlebotomies; until recently, no other pharmacologic therapy could have been considered to modify the iron balance in SCA. With the expanding repertoire of novel therapies that target iron regulatory molecules, new potential therapeutic modalities that do not rely on repeated phlebotomies may finally be tested. Although it is based on a different pathophysiology, a similar therapeutic approach is also being considered for β thalassemia.
Because iron replacement therapy is not totally risk free (see potential increase for malaria infection associated with oral iron supplements), so is the induction of an iron-deficient state. This approach is contra-indicated for young children and pregnant women. Poor adherence to serial phlebotomies has been reported, as well as decreasing fetal HB (Hb F) values with iron restriction. An increased platelet count is a well-known accompanying feature of iron-deficient states, and its pathophysiology is now better understood. Recently, the increased platelet count of iron deficiency anemia has been associated with increased thrombotic risk in a large retrospective survey.10 This potential complication should be carefully considered, given the prothrombotic, procoagulant state of SCD. However, the convergence of multiple case reports, small case series, and the report of Parrow et al in a validated murine model of the disease all seem to indicate that iron restriction should now be seriously considered for both SCA and Hb SC disease, either as repeated phlebotomies or novel therapies, or both.
Conflict-of-interest disclosure: The author declares no competing financial interests.

