

In this issue of Blood, guided by clinical observations and needs, have identified a germline missense mutation in DNA methyltransferase 1 (DNMT1), a ubiquitously expressed key epigenetic regulator, as a cause of hereditary persistence of fetal hemoglobin (HPFH). HPFH protects against β-thalassemia and sickle cell disease (the β-hemoglobinopathies).1 Discussed here is how these findings by Gong et al continue the pioneering role of the β-hemoglobinopathies as a model of discovery for all biomedicine. Sickle cell disease, after all, is the “first molecular disease”: altered migration of sickle vs normal hemoglobin in gel electrophoresis demonstrated, for the first time, that the structure–chemical basis for disease is discoverable and knowable.2
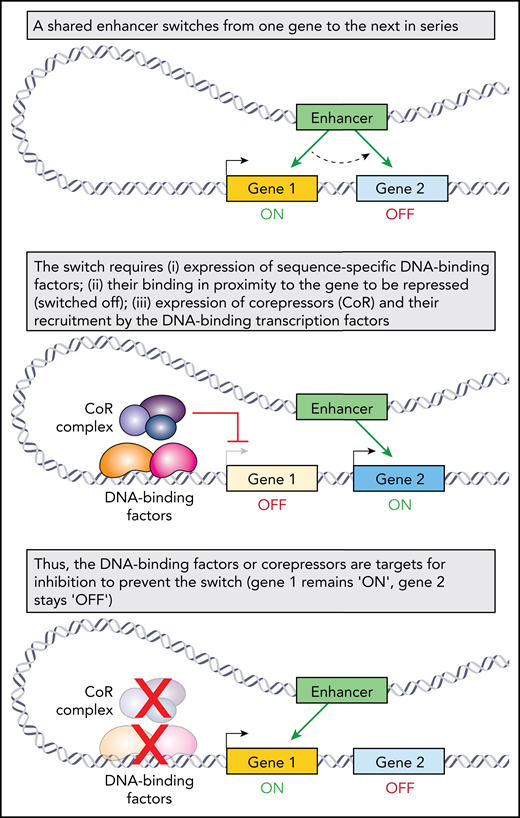
Bedside-to-bench investigation in the β-hemoglobinopathies has pioneered insights into fundamental mechanisms in biology (eg, coordinated, consecutive activation of a gene series). These discoveries have opened the door to rational, noncytotoxic methods of manipulating cell fates and functions (eg, by inhibiting corepressors) for the therapy mission in the β-hemoglobinopathies and beyond.
Bedside-to-bench investigation in the β-hemoglobinopathies has pioneered insights into fundamental mechanisms in biology (eg, coordinated, consecutive activation of a gene series). These discoveries have opened the door to rational, noncytotoxic methods of manipulating cell fates and functions (eg, by inhibiting corepressors) for the therapy mission in the β-hemoglobinopathies and beyond.
The β-hemoglobinopathies become manifest after a switch during development from fetal to adult hemoglobin production. Thus, patients who continue to express high amounts of fetal hemoglobin after infancy receive protection. HPFH describes those individuals with particularly generous fetal hemoglobin amounts (>10%, compared with <1% usually) and correspondingly benign natural histories (reviewed in Thein3 ). Motivated by the goal of using pharmacology to mimic the salutary effects of HPFH, there have long been efforts to dissect the mechanisms involved with the developmental switch from fetal to adult whemoglobin production. The insights gained are fascinating and of general import (see figure). The fetal (HBG2, HBG1), adult (HBB), and other β-globin genes are arrayed together on chromosome 11 (“the β-globin locus”), ordered 5′ to 3′ in the order of their sequential activation by a shared distal (as much as ∼20 kB distant) enhancer, “the locus control region,” that activates each gene in turn.4 In erythroid precursors before 7 months gestational age, this enhancer march from gene to gene stalls at HBG2/HBG1. Above this age, the march proceeds onward to HBB. Germline mutations linked with HPFH have assisted in elucidating the biochemical mechanics of enhancer switching between genes. For example, HPFH-linked point mutations at the locus of BCL11A on chromosome 2, which decreased its expression in the erythroid lineage, implicated this sequence-specific DNA binding factor in switching. In this tradition of discovery, Gong et al identified another HPFH-linked mutation, again not at the β-globin locus, but in DNMT1 on chromosome 19. DNMT1 is famous as the maintenance methyltransferase, that during S-phase, recapitulates the DNA methylation marks (a repression or “off” mark) of the parental strand onto the newly synthesized DNA strand. Notably, DNMT1 is also a corepressor, a protein recruited by sequence-specific DNA binding factors, including BCL11A, to repress, instead of activate, target genes by acting as a platform for other corepressor enzymes and possibly by methylating DNA de novo (reviewed in Gong et al and Molokie et al5 ).
Several facets of this discovery are worthy of attention and discussion. (1) The discovery was not from population-based genome-wide association study. Rather, the investigators used next generation sequencing, complemented by Sanger sequencing as needed, to examine exons and presumed regulatory regions of a shortlist of 49 genes in 1142 β-thalassemia patients. These 49 genes were already implicated in regulation of the β-globin locus. This approach increased their power to identify rare variants in these candidate genes that might explain some instances of HPFH (∼50% of fetal hemoglobin variation in adult humanity remains unexplained3 ). They found the DNMT1 mutation in 3 of the 1142 patients, all 3 of whom did have substantially elevated fetal hemoglobin levels of between ∼32% and 50%. (2) The mutation they discovered is missense, c.2633G>A, S878F. That is, a nucleophilic serine is substituted with an aromatic phenylalanine; the substitution occurs in a bromo-adjacent homology domain of DNMT1 (not in the methyltransferase catalytic domain), which does influence DNMT1 protein folding in structural studies (other functions are unknown) (reviewed in Jeltsch and Jurkowska6 ). The authors moreover showed that the substituted serine can be phosphorylated, a posttranslational modification they correlated with increased DNMT1 protein stability; the substitution hence correlated with decreased protein stability. They also found decreased interactions of mutated DNMT1 S878F with BCL11A and other proteins. (3) Better natural histories of β-hemoglobinopathy patients with co-inherited HPFH implies that the phenotypic consequences of HPFH mutations, present in every cell of the body, are restricted to the erythroid lineage. Accordingly, HPFH-linked BCL11A mutations are not in BCL11A protein-coding regions but in regulatory elements that drive erythroid-specific gene activation.7 HPFH-linked KLF1 mutations do disrupt the DNA-binding domain of this key transcription factor, but KLF1 expression and function is inherently restricted to the hematopoietic lineage (reviewed in Borg et al8 ). How then to explain HPFH-linked mutation in DNMT1, ubiquitously expressed and with fundamental functions as the maintenance methyltransferase and as a corepressor? One possibility (the only possibility?) is that S878 phosphorylation is an erythroid-lineage-specific posttranslation modification, explaining the selection for DNMT1 c.2633G>A, S878F in β-thalassemia pedigrees. Certainly, the logical next step is to investigate DNMT1 S878 phosphorylation further. (4) This discovery supports a model in which switching of a shared enhancer from 1 gene to another, likely a generalizable motif for regulated consecutive expression of gene series, requires that the presently activated gene is silenced first3 (see figure). (5) Finally, the discovery aligns with active clinical and preclinical efforts using small molecules that inhibit/deplete DNMT1 to upregulate fetal hemoglobin.5,9 The discovery also implicates the kinase-mediating phosphorylation of DNMT1 S878 as a candidate for inhibition as a potential new avenue to pharmacologic recapitulation of HPFH.
In sum, this discovery indicates that posttranslational modification of a key corepressor, by a kinase-based pathway that is amenable to sensitive command and control, is a method by which consecutive activation of a gene series is regulated. Highlighting the broader implications for biomedicine, clinical DNMT1-targeting is already approved for myeloid tumor therapy, is in evaluation for treatment of several other cancers, does prevent a switch from fetal to adult hemoglobin production in the erythroid lineage, and does affect master transcription factor “switching” to redirect lineage-fate trajectories (reviewed in Velcheti et al10 ). The β-hemoglobinopathies thus continue their tradition of revealing life’s mechanisms for all biomedicine.
Conflict-of-interest disclosure: Y.S. has issued patents around tetrahydrouridine and decitabine, ISWI family inhibition, and cancer differentiation inducers, and has equity, consulting, and Board interest in EpiDestiny, which has licensed oral tetrahydrouridine-decitabine.


