In this issue of Blood, provide compelling evidence that the stabilization of transcription factor hypoxia-inducible factor 2 (HIF2) promotes the motility of polymorphonuclear leukocytes (PMN), especially neutrophils, through highly constrained matrices.1
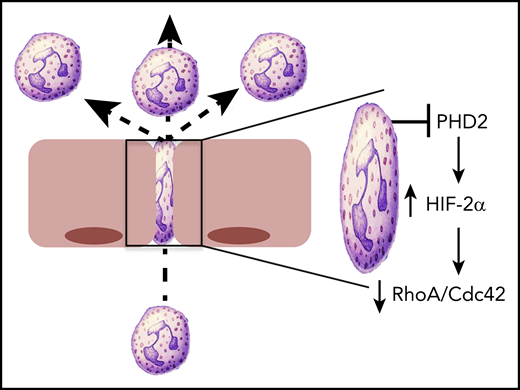
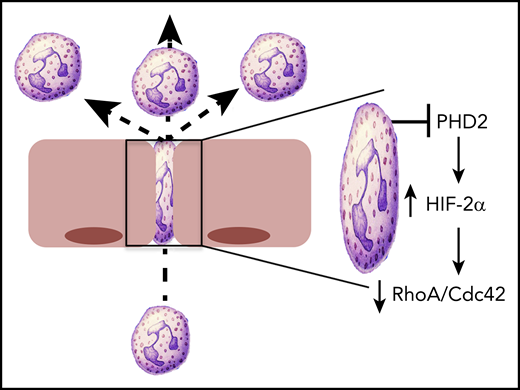
HIF2 regulates PMN motility. Conditional deletion of PHD2 in PMN results in the stabilization of HIF2α and the attenuation of cytoskeletal activity, mediated by loss of RhoA and Cdc42. This loss of cytoskeletal activity was particularly evident in the constrained environment that would be present during PMN transmigration, with resulting increases in PMN motility.
HIF2 regulates PMN motility. Conditional deletion of PHD2 in PMN results in the stabilization of HIF2α and the attenuation of cytoskeletal activity, mediated by loss of RhoA and Cdc42. This loss of cytoskeletal activity was particularly evident in the constrained environment that would be present during PMN transmigration, with resulting increases in PMN motility.
Acute inflammatory responses are characterized by the accumulation of large numbers of neutrophils. The migration of such neutrophils to sites of infection or injury is accompanied by energy-demanding processes such as phagocytosis and the generation of reactive oxygen intermediates. These energy-taxing activities have the potential to shift the metabolic homeostasis of inflamed tissues toward significant hypoxia and activation of HIF.2
HIF is considered the central regulator of hypoxia adaptation. Discovered in 1992 on the basis of its ability to regulate erythropoietin, it was the topic of the 2019 Nobel Prize in Physiology or Medicine.3 HIF1α was the original isoform purified by oligonucleotide binding to the 3′ region of the EPO gene.4 HIF2α was subsequently identified by homology searches as a binding partner for the heterodimeric partner HIF1β.5 HIF3α is a more distantly related isoform, and when spliced appropriately, it can encode a protein that antagonizes hypoxia-responsive element (HRE)-dependent gene induction.6 The stabilization of the α subunit of HIF is dependent on any combination of HIF-selective iron and O2-dependent prolyl hydroxylase domain protein 1 (PHD1), PHD2, and PHD3, which hydroxylate prolines 564 and 402 within the O2-dependent degradation domain of the HIFα subunit.7,8 By using conditional gene deletion strategies, Sormendi et al demonstrated that PHD2 deficiency prominently stabilizes HIF2 in murine PMN. By using an elegant system of 1-dimensional (1D) and 2D migration assays engineered to allow for analysis on a constrained substrate, they showed that HIF2 stabilization significantly promotes the speed of PMN migration in confined spaces. The authors used an engineered micro-constriction device to show that PHD2-deficient PMN display enhanced motility in confined 3D spaces, modeling the environment a PMN may experience within tight vascular or parenchymal spaces within tissues (see figure). Extensions of these studies in vivo using intravital microscopy in the mouse ear revealed that loss of PHD2 enhanced the speed of PMN transendothelial migration and increased the recruitment of PMN into the synovium in a serum-induced inflammatory arthritis model.
To gain insight into mechanisms of enhanced PMN migration, Sormendi et al performed deep RNA sequencing that compared PHD2-deficient PMN with their wild-type counterparts. This analysis revealed the predictable glycolysis and gluconeogenic phenotype associated with HIF-regulated metabolism and also identified a prominent cell cytoskeletal signature, in particular the loss of 2 small Rho GTPases, RhoA and Cdc42 (see figure). The protein-protein interaction network map revealed nearly 50 interactive components between RhoA and Cdc42. The Rho family of GTPases functions as molecular switches by cycling between the GTP-bound active form and the GDP inactive form. In their active state, Rho family members play key roles in all aspects of cytoskeletal activity and reorganization. In particular, RhoA mediates actin turnover via its association with Rho-associated protein kinase (ROCK) and actin polymerization through its association with the formin family of proteins (eg, mDia). The finding that loss of RhoA enhances motility is particularly interesting. The major function of RhoA in PMN chemotaxis was originally thought to be in tail retraction during migration. This idea was subsequently disproved by Jennings et al9 who used RhoA-deficient PMN to demonstrate increased chemokinesis and chemotaxis, an observation akin to those in the Sormendi et al study. It is also noteworthy that decreased RhoA activity may be associated with loss of PI3-kinase activity,10 the latter representing one of the major braking mechanisms in PMN migration11 that is strongly tied to HIF-mediated signaling.12
Given the significant interest in the development of therapeutics around the HIF pathway, it is tempting to speculate whether this work may provide a rationale for the treatment of inflammatory disorders. Substantial efforts have gone into the development of PHD inhibitors (ie, HIF stabilizing agents).13 On the basis of the work by Sormendi et al, these inhibitors could promote PMN migration and potentially enhance bacterial clearance in immune suppressed individuals. Conversely, HIF inhibitors have also come to light. For example, PT2977 is a novel oral HIF2α inhibitor14 that could be imagined as an inhibitor of PMN migration in diseases in which PMN might cause bystander tissue damage. The answer to whether such applications might prove beneficial in patients with inflammatory disorders is eagerly anticipated.