

In this issue of Blood, identified the mechanism of excessive complement activation caused by recurrent mutations in factor H–related protein-1 (FHR-1) in a few patients with atypical hemolytic uremic syndrome (aHUS). These are rare mutations in a rare disease, but the results provide a better understanding of complement regulation.1
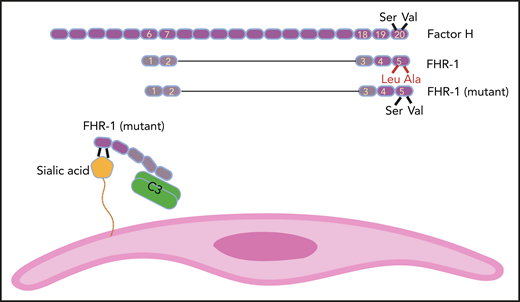
FH has 20 SCR domains, and FHR-1 has 5 of them. SCR domains of FHR-1 and FH are homologs, but FHR-1 does not possess complement regulatory domains SCR1 to 4 of FH. SCR 5 of FHR-1 is almost identical to SCR 20 of FH, except for 2 amino acids (Leu290 and Ala296 in FHR-1 instead of Ser1191 and Val1197 in FH). In some aHUS patients, these amino acids in FHR-1 are mutated back to their FH counterparts. As a result, mutant FHR-1 can bind with a higher affinity to sialic acid residues on the cell surface (eg, endothelial cells). Mutant FHR-1 molecules also bind C3 and bring it close to the cell surface, which promotes complement activation.
FH has 20 SCR domains, and FHR-1 has 5 of them. SCR domains of FHR-1 and FH are homologs, but FHR-1 does not possess complement regulatory domains SCR1 to 4 of FH. SCR 5 of FHR-1 is almost identical to SCR 20 of FH, except for 2 amino acids (Leu290 and Ala296 in FHR-1 instead of Ser1191 and Val1197 in FH). In some aHUS patients, these amino acids in FHR-1 are mutated back to their FH counterparts. As a result, mutant FHR-1 can bind with a higher affinity to sialic acid residues on the cell surface (eg, endothelial cells). Mutant FHR-1 molecules also bind C3 and bring it close to the cell surface, which promotes complement activation.
aHUS is a systemic thrombotic microangiopathy that results from activation of the alternative complement pathway thus causing endothelial injury. Mutations in genes encoding complement or complement regulatory proteins can be identified in about half of aHUS patients. Most of the detected mutations are in the complement factor H (CFH) gene, and most of the mutations in CFH affect the C-terminal regions of factor H (FH). FH is a plasma protein that inhibits the alternative complement pathway.2 It is a cofactor for factor I–mediated degradation of C3b on the cell surface and also in plasma (cofactor activity), and it can dissolve C3 and C5 convertases (decay-accelerating activity).3 To degrade C3b on the cell surface, FH needs to perform complicated molecular gymnastics and to bind to surface polyanions (glycosaminoglycans, heparan, and sialic acid) and C3b at the same time. This flexibility is possible because of a unique structural arrangement. FH comprises 20 homologous subunits that, like beads on a string, give FH the ability to fold back on itself.4 Each subunit, also known as the short consensus repeat (SCR) domain or sushi domain, has about 60 amino acids. Despite structural similarities, there are functional distinctions between different SCR domains of FH. The SCRs in the N-terminus of FH (SCRs 1-4) have complement regulatory activity, and the SCRs in the C-terminus (SCRs 18-20) are essential for adhesion to the cell surface by binding to polyanions and C3b. Mutations in the C-terminal SCR domains (particularly SCRs 19 and 20) or antibodies to these subunits prevent FH from binding to cells and cause aHUS or membranoproliferative glomerulonephritis. FH mutants can inhibit complement activation in plasma but cannot bind to the cell surface to prevent complement activation on cells, resulting in complement-induced damage.
FH is a member of the FH family proteins that also includes FHR-1, FHR-2, FHR-3, FHR-4, and FHR-5. FHR proteins have between 4 and 9 SCRs, and their common feature is the loss of the N-terminal complement regulatory activity that in FH is mediated by SCRs 1 to 4 (see figure). On the other end, C-terminal SCRs of FHR proteins keep their homology to C-terminal SCRs of FH. For example, FHR-1 has 5 SCRs, and its SCRs 3 and 4 are identical to FH SCRs 18 and 19. The SCR 5 of FHR-1 is 97% similar to SCR 20 of FH and is different only in 2 amino acids (Leu290 and Ala296 in FHR-1 instead of Ser1191 and Val1197 in FH).5 As a result, FHR-1 can bind to deposited C3b on the cell surface but does not inhibit complement activation (complement deregulation).6
The structural similarity and functional difference between FH and FHR-1 raise the possibility that competition between FHR-1 and FH for binding to deposited C3b on the cell surface may play a role in the pathogenies of complement disorders (age-related macular degeneration, aHUS, C3 glomerulopathy, and immunoglobulin-A–associated nephropathy).7,8
The authors previously identified 2 mutations in SCR 5 of FHR-1 (L290S and A296V) in 9 of 531 aHUS patients.9 These mutations are probably caused by the transfer of sequence from CFH to CFHR-1 (gene conversion), aided by their proximity in the regulators of complement activation gene cluster on chromosome 1. These mutations make SCR 5 of FHR-1 identical to SCR 20 of FH. In the Martin Merinero et al study, the authors extended their investigation of how the enhanced resemblance of mutant FHR-1 to FH can be pathogenic.
In a series of functional studies, using traditional complement biology tools (such as sheep erythrocyte lysis assay), innovative experiments (such as incubation of kidney sections from CFHR-1– and CFH-deficient mice with serum), and elegant structural studies that used nuclear magnetic resonance, the authors showed that mutant FHR-1 has a higher affinity for sialic acid than wild-type FHR-1. This finding by itself could not explain the pathogenicity of mutant FHR-1 because FH has a higher plasma concentration than FHR-1, and the copious amount of sialic acid on the cell surface can accommodate both FH and FHR-1.
To complete the picture, the authors showed that mutant FHR-1 could also bind with a higher affinity to C3. As a result of binding to both cell surface and plasma C3, one can speculate that mutant FHR-1 molecules bring C3 into close juxtaposition to the cell thus providing suitable conditions for complement activation on the cell surface. This last speculation needs to be further investigated. The Martin Merinero et al study provides an important perspective on the function of FH family proteins and shows that the complement system still has many surprises that are yet to be discovered.
Conflict-of-interest disclosure: The author declares no competing financial interests.

