Key Points
Nivolumab monotherapy was associated with very limited activity in patients with R/R FL.
Understanding the tumor microenvironment may be necessary to develop effective checkpoint-based strategies in FL.

Abstract
Nivolumab, an anti–programmed death-1 (PD-1) monoclonal antibody, showed promising activity in relapsed or refractory (R/R) follicular lymphoma (FL) in a phase 1 study. We conducted a phase 2 trial to further evaluate its efficacy and safety in patients with R/R FL and to explore biomarkers of response. Patients with R/R FL and at least 2 prior lines of therapy, each containing a CD20 antibody or an alkylating agent, were treated with nivolumab 3 mg/kg every 2 weeks. The primary end point was objective response rate (ORR) assessed by an independent radiologic review committee. Biomarker analyses included gene expression profiling and multiplex immunofluorescence studies of pretreatment tumor samples. A total of 92 patients were treated. After a minimum follow-up of 12 months, ORR was 4% (4 of 92 patients). Median progression-free survival (PFS) was 2.2 months (95% confidence interval [CI], 1.9-3.6 months). Median duration of response was 11 months (95% CI, 8-14 months). Exploratory analyses suggested that responders had significantly higher proportion of CD3+ T cells in the tumor microenvironment than nonresponders, but no significant differences in PD-1 or programmed death-ligand 1 expression were observed. High expression of a set of tumor-associated macrophage genes was associated with reduced PFS (hazard ratio, 3.28; 95% CI, 1.76-6.11; P = .001). The safety profile was consistent with previous reports of nivolumab. In conclusion, nivolumab monotherapy was associated with very limited activity in patients with R/R FL. Better understanding of the immune biology of this disease may facilitate the development of effective checkpoint-based strategies. This trial was registered at www.clinicaltrials.gov as #NCT02038946.
Introduction
Follicular lymphoma (FL) typically has an indolent disease course, but is not considered curable with conventional therapies.1 Most patients experience relapse after frontline therapy, and although newer therapies such as PI3K inhibitors and the immunomodulatory drug lenalidomide may lead to frequent responses in patients with relapsed or refractory (R/R) disease, most patients will subsequently relapse.2-5 Therefore there is a need for novel, effective, and safe therapies for R/R FL.
FL appears to be inherently immunosensitive, as evidenced by the success of allogeneic hematopoietic cell transplantation (HCT),6,7 as well as occasional responsiveness to other immunotherapeutic agents such as ipilimumab and pidilizumab.8,9 Nivolumab, a fully human IgG4 monoclonal antibody against programmed death-1 (PD-1), blocks signaling induced by the binding of PD-1 to its ligands programmed death-ligand 1/ligand 2 (PD-L1/L2), which releases inhibition of T cells and restores antitumor immune responses.10 Nivolumab is indicated for the treatment of several tumor types.10 In CheckMate 039, a phase 1 study targeting several lymphoid malignancies, nivolumab showed promising activity in R/R FL, with an objective response rate (ORR) of 40% (4 of 10 patients).11 Responses were durable, with response durations >1.5 years for 1 patient with complete remission (CR) and ≥6 months for patients with partial remission (PR). This study also suggested a favorable safety profile for nivolumab in R/R FL, similar to that observed in patients with solid tumors and classical Hodgkin lymphoma.12 On the basis of these encouraging results, we conducted a phase 2 study to evaluate the efficacy and safety of nivolumab in patients with R/R FL and previous rituximab treatment.
The mechanistic basis of responsiveness to PD-1 blockade in FL has yet to be elucidated. Most FL tumor cells do not express PD-L1/L2,11,13-15 but PD-L1 has been found on nonmalignant tumor-infiltrating immune cells.11,15 Meanwhile, tumor-infiltrating T cells showed high PD-1 expression and suppressed cytokine signaling.15 Thus, responses to nivolumab could provide important insights into the mechanism of PD-1 blockade in FL. Indeed, the host immune response has been recognized as an important determinant of sensitivity of FL to conventional therapy. In 1 study, 2 gene expression signatures of nonmalignant tumor-infiltrating immune cells, termed immune-response 1 (IR-1) and immune-response 2 (IR-2), were associated with favorable and poor survival prognoses in FL, respectively.16 Studies have also found that the degree of infiltrating macrophages and T cells may be prognostic in FL,17 and progression of disease within 24 months was associated with reduced intratumoral immune infiltration.18 In this study, therefore, we examined pretreatment biomarkers as potential predictors of response to nivolumab.
Methods
Study design and patient population
CheckMate 140 (NCT02038946) was a single-arm, open-label, phase 2 trial in patients aged ≥18 years with R/R FL after failure of at least 2 prior lines of therapy, each of which had to contain a CD20 antibody or an alkylating agent, and at least 1 had to include rituximab. Patients were excluded if they had central nervous system involvement, prior treatment with antibodies targeting T-cell costimulation or immune checkpoint pathways, or prior allogeneic HCT or autologous HCT within 12 weeks prior to receiving study drug. The study was conducted in accordance with good clinical practice, as defined by the International Conference on Harmonization. The study protocol, patient consent forms, and patient recruitment material were approved by the institutional review board/independent ethics committee at each participating study center and conducted in accordance with the Declaration of Helsinki. All patients provided written informed consent before study initiation. The original protocol contained an interim analysis for futility; however, since further follow-up of the phase 1 CheckMate 039 study showed that many responses were delayed, with half occurring around 9 months,11 this futility analysis was removed.
Treatment
Patients were treated with 3 mg/kg nivolumab every 2 weeks until disease progression, unacceptable toxicity, or withdrawal of consent. Nivolumab was administered as an IV infusion over 60 minutes on day 1 of each treatment cycle. Patients still meeting all other eligibility criteria could continue to receive nivolumab beyond progression if they were deriving apparent clinical benefit from treatment in the opinion of the treating investigator.
Assessments and end points
Tumor responses were assessed by computed tomography or magnetic resonance imaging at baseline, every 8 weeks through the first 8 months, every 12 weeks through months 9 to 24, and every 6 months thereafter until disease progression. Positron emission tomography scans were required at baseline and to confirm a CR. Bone marrow biopsy or aspirate was required within 90 days prior to consent or during the screening period. For patients with bone marrow involvement at screening, bone marrow biopsy or aspirate was required to confirm a CR. A minimum of 1 formalin-fixed paraffin-embedded (FFPE) tumor tissue block or minimum of 10 FFPE unstained sections were required for biomarker evaluations.
The primary end point was ORR assessed by independent radiologic review committee (IRC) according to 2007 International Working Group Response Criteria.19 The final analysis of the primary end point occurred 12 months after the last enrolled patient’s first dose of nivolumab. Secondary end points included duration of response (DOR), CR rate, PR rate, and progression-free survival (PFS), all assessed by IRC, and ORR by investigator assessment. Exploratory end points included safety and tolerability, overall survival (OS), and the association of biomarkers with efficacy measures. Specific biomarkers for evaluation were selected post hoc.
Biomarker assessment
Expression of CD3, CD68, PD-1, and PD-L1 in the tumor microenvironment was analyzed by multispectral immunofluorescence from a subsample of responding and nonresponding patients. A pretreatment biopsy was chosen for biomarker studies when possible, and archival tissue was used if pretreatment tissue was insufficient. Staining was performed using BOND RX fully automated stainers (Leica Biosystems, Wetzlar, Germany). Image acquisition was performed using the Mantra multispectral imaging platform (PerkinElmer, Waltham, MA). Areas with nontumor or residual normal tissue were excluded from the analysis. Representative regions of interest were chosen by the pathologist, and multiple fields of view were acquired at ×20 resolution as multispectral images. Cell identification was performed using supervised machine learning algorithms within Inform 2.4 (PerkinElmer). Thresholds for staining and the accuracy of phenotypic algorithms were optimized and confirmed by the pathologist for each case.
Gene expression profiling on FFPE pretreatment tumor specimens was performed using the HTG EdgeSeq oncology biomarker panel (HTG Molecular Diagnostics, Inc., Tucson, AZ). Raw counts for the 2568 genes on the panel were normalized using total counts; a negative binomial was used to model the count distribution using EdgeR software.
RNA sequencing was performed using pretreatment tumor specimens for expression profiling of the complete set of genes in the IR-1 and IR-2 signatures16 (except for NDN and CEB1 from IR-2) in a select group of responders and nonresponders. Data analysis was performed using the Wilcoxon rank-sum test.
Levels of cytokines were measured using Luminex-based technology (HumanMAP panel; Myriad RBM, Austin, TX) from serum collected at baseline and select time points during treatment. Differences in the sum of ranks between groups were compared using Dunn’s multiple comparisons test.
Statistical analysis
The desired sample size of 90 patients was chosen to provide 87% power to reject the null hypothesis that the ORR is ≤20%, with a 2-sided α of 5%. This sample size was also considered sufficient for the safety analysis. All patients who received at least 1 dose of nivolumab were included in the efficacy and safety analyses. ORR by IRC was summarized by a binomial response rate and its corresponding 2-sided 95% exact confidence intervals (CI) using the Clopper–Pearson method. CR and PR rates by IRC, and ORR by investigator were summarized similarly. DOR by IRC, PFS by IRC and investigator, and OS were summarized based on the Kaplan-Meier product-limit method. Median values of PFS with 2-sided 95% CIs (based on the log-log transformation) were calculated. Safety analyses were performed using descriptive statistics using NCI CTCAE version 4.0.
Results
Baseline characteristics and patient disposition
A total of 92 patients were treated with nivolumab. Baseline clinical characteristics of all treated patients are shown in Table 1. Median number of prior therapies was 3 (range, 2-10 therapies), with 42% of patients having at least 4 prior therapies. The percentages of patients with R/R disease at baseline were 65% and 35%, respectively, and 45% of patients had progression of disease within 24 months. At the clinical cutoff, the minimum follow-up was 12 months. A total of 87 patients (95% of all treated) discontinued study treatment. Reasons for discontinuation were disease progression (76%), study drug toxicity (9%), patient request/withdrawal of consent (7%), adverse events (AEs) unrelated to study drug (6%), lost to follow-up (1%), and no longer meeting study criteria (1%). The median number of doses received was 8 (range, 1-50 doses).
Safety
In total, 95% of patients experienced at least 1 AE and 49% experienced at least 1 grade 3-4 AE. The most common grade 3-4 AEs were malignant neoplasm progression (5%), neutropenia (5%), abdominal pain (4%), and anemia (4%). Treatment-related AEs (TRAEs) occurred in 54% of patients, most frequently fatigue (13%), diarrhea (11%), and nausea (10%) (Table 2). Most TRAEs were grade 1-2; 15% of patients experienced at least 1 grade 3-4 TRAE, but no grade 3-4 event occurred in more than 2% of patients (Table 2). Serious AEs (SAEs) were reported in 39% of patients. The most frequent SAEs were malignant neoplasm progression (5%; all were grade 3-4) and pyrexia (4%; all were grade 1-2). Immune-related AEs included rash (9%), diarrhea/colitis (2%), hypersensitivity/infusion reactions (2%), hepatitis (1%), and pneumonitis (1%) (supplemental Table 1, available on the Blood Web site). Three patients discontinued treatment because of immune-mediated AEs (pneumonitis, rash, and toxic epidermal necrolysis; 1 patient each).
In all, 30 patients (33%) died during the study, most commonly because of disease progression (17 patients [18%]). There were 3 deaths (3%) related to study drug toxicity: 1 patient with toxic epidermal necrolysis, 1 with acute respiratory failure and septic shock, and 1 with erythema multiforme.
Efficacy
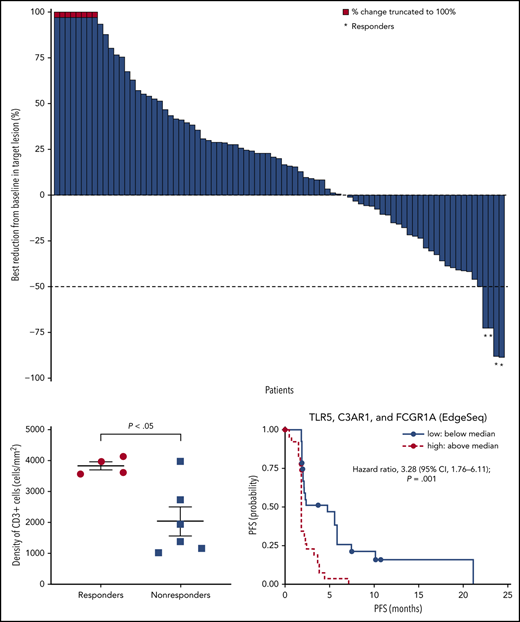
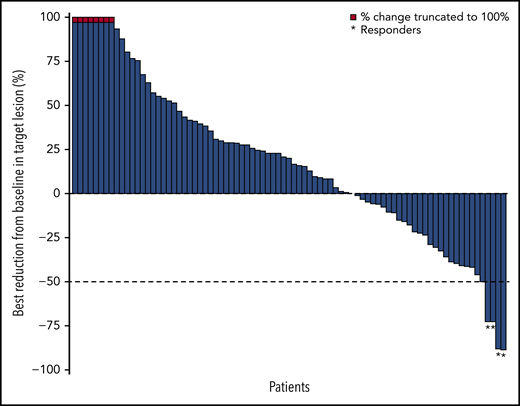
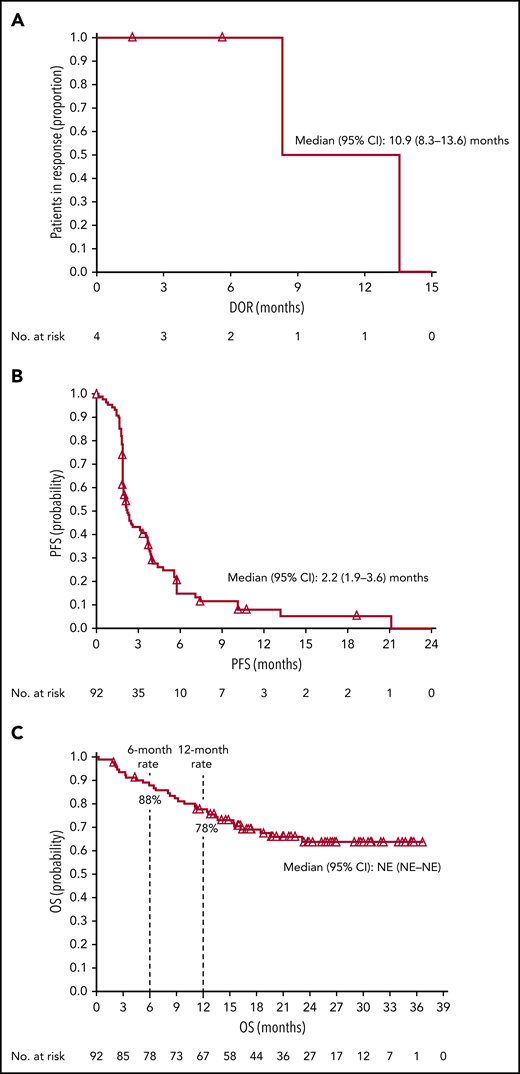
The ORR based on IRC assessment was 4% (4 of 92 patients); 1 patient achieved CR and 3 patients achieved PR (Table 3). The investigator-assessed ORR was 11% (10 of 92 patients), with 2 CR and 8 PR. More than one-third (36%) of evaluable patients experienced reductions in target lesion burden (Figure 1). For patients who responded to treatment, median time to response was 5.0 months (interquartile range, 2.1-10.4 months), and the median DOR was 10.9 months (95% CI, 8.3-13.6 months) (Figure 2A). Median time to next treatment was 5.9 months (95% CI, 4.3-6.6 months). Median PFS per IRC was 2.2 months (95% CI, 1.9-3.6 months) (Figure 2B). PFS was similar when stratified by number of prior lines of therapy; median PFS was 3.3 months, 2.1 months, and 2.0 months in patients with 2, 3, or ≥4 prior lines of therapy, respectively. Median PFS was also similar when stratified by best overall response to most recent prior therapy: 2.3 months among patients with CR, 3.7 months with PR, 2.3 months with stable disease (SD), and 1.9 months with progressive disease. The 6-month and 12-month OS rates were 88% (95% CI, 79-93%) and 78% (95% CI, 68-85%), respectively (Figure 2C). The median OS had not been reached by the clinical cutoff date.
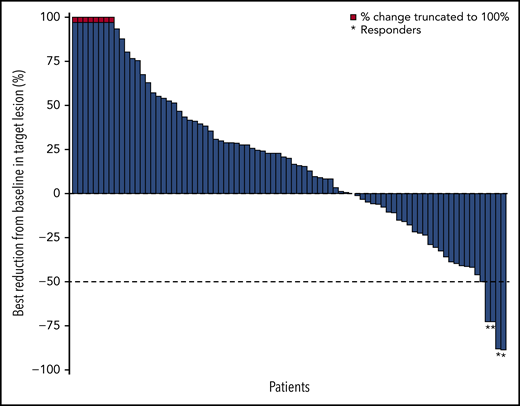
Best change in target lesions in response-evaluable patients per IRC. Response-evaluable patients (n = 83) were those with a target lesion(s) assessed at baseline and with at least 1 on-study time point with all baseline target lesion(s) assessed. Negative values indicate maximum tumor reduction; positive values indicate minimum tumor increase; best change is based on evaluable target lesion measurements up to progression or start of subsequent therapy. Dashed horizontal line indicates the 50% reduction consistent with a response per revised 2007 International Working Group criteria.
Best change in target lesions in response-evaluable patients per IRC. Response-evaluable patients (n = 83) were those with a target lesion(s) assessed at baseline and with at least 1 on-study time point with all baseline target lesion(s) assessed. Negative values indicate maximum tumor reduction; positive values indicate minimum tumor increase; best change is based on evaluable target lesion measurements up to progression or start of subsequent therapy. Dashed horizontal line indicates the 50% reduction consistent with a response per revised 2007 International Working Group criteria.

Kaplan-Meier estimates. (A) DOR per IRC, (B) PFS per IRC, and (C) OS. Open triangles represent censored observations. NE, not estimable.
Kaplan-Meier estimates. (A) DOR per IRC, (B) PFS per IRC, and (C) OS. Open triangles represent censored observations. NE, not estimable.
Biomarkers
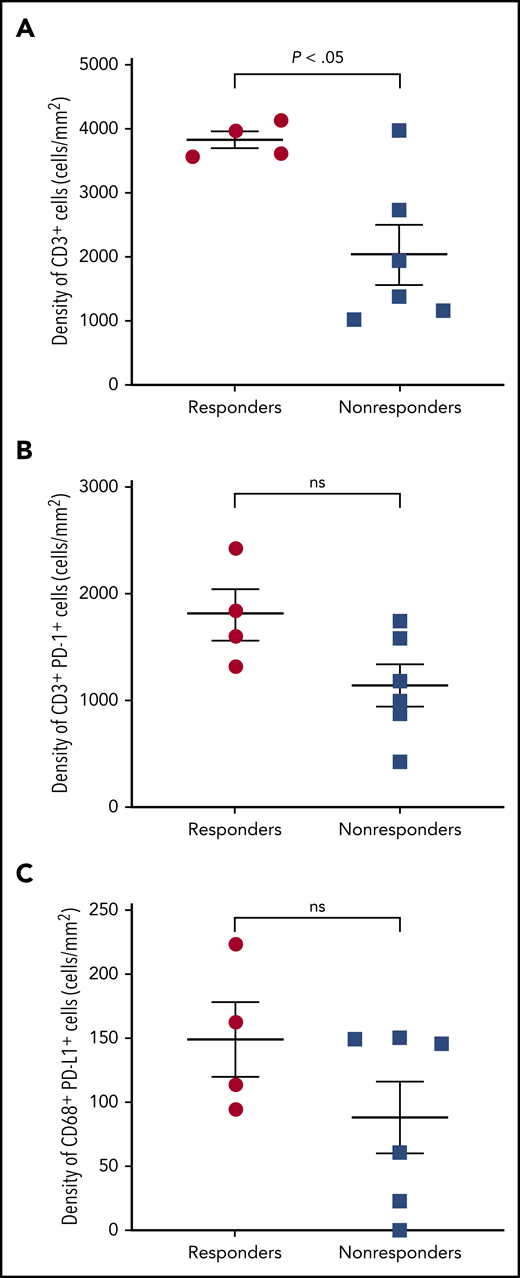
Selected tumor specimens (n = 10) from 4 responders and 6 nonresponders (SD or progression) per investigator were evaluated by multispectral immunofluorescence. Significantly more infiltrating CD3+ T cells were observed in FL specimens from responders vs nonresponders (P = .016; Figure 3A). Most PD-1 was expressed on CD3+ T cells and most PD-L1 on CD68+ macrophages (not shown). More tumor-infiltrating CD3+PD-1+ T cells and CD68+PD-L1+ macrophages were observed in responders than nonresponders, but these differences were not statistically significant (Figure 3B-C).
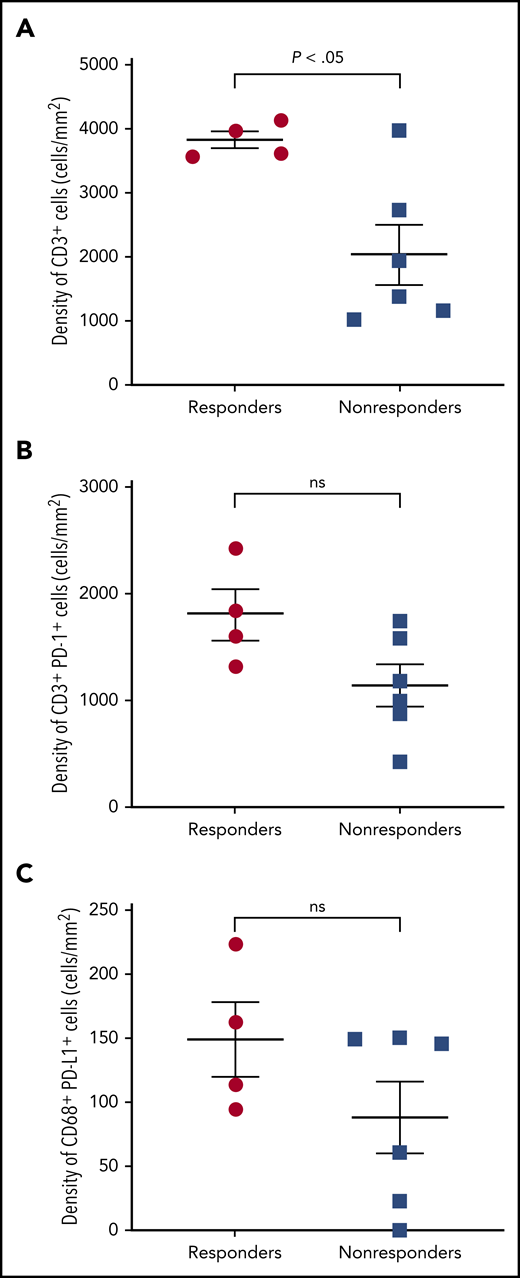
Multispectral immunofluorescence. (A) CD3, (B) CD3 and PD-1, and (C) CD68 and PD-L1 in patients with and without response to nivolumab. ns, not significant.
Multispectral immunofluorescence. (A) CD3, (B) CD3 and PD-1, and (C) CD68 and PD-L1 in patients with and without response to nivolumab. ns, not significant.
In analyses of pretreatment serum levels of 49 cytokines among 78 patients, soluble interleukin 2 receptor α (sIL-2Rα) levels were lower at baseline in responders per investigator vs nonresponders (supplemental Figure 1) as assessed by rank-sum testing (P = .0025), though the absolute differences were small.
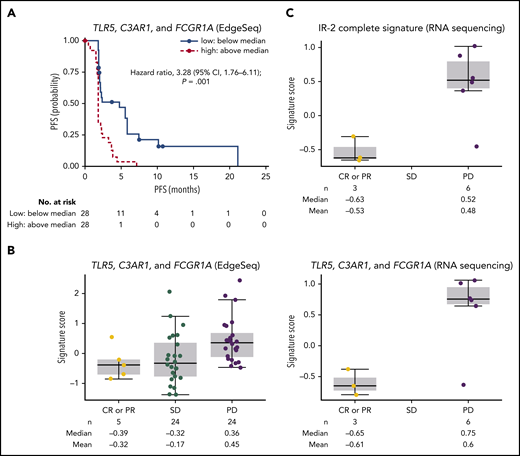
A total of 56 patients had evaluable gene expression profile data for HTG EdgeSeq oncology biomarker panel, and 11 patients for RNASeq. Among an extensive list of published gene signatures involved in immune responses, an IR-2 panel comprising select genes from the IR-2 signature16 was strongly associated with PFS (supplemental Figure 2). We also assessed the association between PFS and the expression of specific tumor-associated macrophage genes within the IR-2 signature (TLR5, C3AR1, and FCGR1A). The risk of disease progression or death was significantly increased in patients with high (above median) levels of baseline TLR5, C3AR1, and FCGR1A vs patients with low (below median) levels, with a hazard ratio of 3.28 (95% CI, 1.76-6.11; P = .001) (Figure 4A). Median PFS was 1.9 months (95% CI, 1.7-2.1) and 4.8 months (95% CI, 2.0-5.8) for patients with high and low baseline levels of TLR5, C4AR1, and FCGR1A, respectively. Levels of baseline TLR5, CA4AR1, and FCGR1A (as assessed by signature score) were higher in patients with progressive disease compared with patients who achieved CR or PR per investigator (Figure 4B). The correlation between expression level and best overall response was also significant (P = .048) when the complete set of genes in the IR-2 signature was included in the analysis, with a median signature score of −0.63 in patients with CR or PR, and 0.52 in patients with progressive disease (Figure 4C). In contrast, no significant association (P = .56) was observed between expression levels of a group of genes within the IR-1 signature16 expressed in T cells (TNFSF13B, LEF1, and STAT4) and PFS. Median PFS was 2.1 months (95% CI, 1.8-2.3) and 2.3 months (95% CI, 1.9-4.4) for patients with high and low levels of these genes at baseline.

Outcomes by gene signatures. (A) PFS per IRC by TLR5, C3AR1, and FCGR1A expression. (B) Best objective response per investigator by TLR5, C3AR1, and GCGR1A score. (C) Best objective response by IR-2 complete signature score. PD, progressive disease.
Outcomes by gene signatures. (A) PFS per IRC by TLR5, C3AR1, and FCGR1A expression. (B) Best objective response per investigator by TLR5, C3AR1, and GCGR1A score. (C) Best objective response by IR-2 complete signature score. PD, progressive disease.
Discussion
The safety profile of nivolumab in patients with R/R FL was similar to that in other settings, although it is notable that 2 fatal dermatologic complications occurred. Although severe dermatologic toxicities have been previously described with PD-1 blockade,20 these are rare events; future studies with PD-1 blockade in FL could therefore pay close attention to this type of toxicity. Unlike the results in the phase 1 study of nivolumab in R/R FL, however, the efficacy of nivolumab monotherapy in this disease was very limited, with an IRC-assessed ORR of 4%, which did not meet the primary end point. As in other settings with PD-1 blockade,12 the investigator-assessed ORR (11%) was higher than IRC-assessed ORR, and the results per investigator were generally consistent with those per IRC. The difference between the present results and those of the phase 1 study may reflect the larger sample size in the phase 2 study.11 Our results also differ from interim results of a phase 2 trial evaluating pembrolizumab in combination with rituximab in patients with R/R FL, in which the ORR was 64%.21,22 In that trial, patients had to be sensitive to rituximab and were less heavily pretreated, with a median of 1 prior line of therapy. Further analyses will be necessary to determine whether the difference in patient populations explains the difference in efficacy or whether there is indeed synergy between PD-1 blockade and rituximab.
Although responses were rare with nivolumab, they appeared durable, consistent with findings from the phase 1 study and prior study of CTLA-4 blockade.8 To understand the biological basis of such prolonged response, which could yield valuable insights into the immune biology of FL and lead to the rational design of effective combination therapies, we examined potential predictive biomarkers in exploratory post hoc analyses. In previous studies, PD-1 was found to be highly expressed on the intratumoral CD4+ follicular T helper cells in the lymph node follicles, and also expressed on exhausted CD4+ cells at lower levels.15,23 Because FL tumors typically lack expression of the PD-1 ligands,11 it is unlikely that direct disruption of a PD-L1/PD-1 interaction between tumor and T cells underlies responses to PD-1 blockade in FL. In our study, PD-L1 was present on CD68+ macrophages, and CD3+ T cells were more abundant in responding patients than nonresponders. A slightly higher proportion of CD3+PD-1+ and CD68+PD-L1+ cells was found in responders than nonresponders, but given the small sample size, the validity of this observation will need to be confirmed. The higher responsiveness of more inflamed tumors has been well described with PD-1 blockade.24,25 Recent work in FL suggested that a population of PD-1+ γδ T lymphocytes may provide a potential source of tumor control that is impaired by PD-1 expression and, therefore, could be recruited by PD-1 blockade.26 There is also evidence that other immune checkpoints are expressed in the FL microenvironment including lymphocyte-activation gene 3 (LAG-3),27 T-cell immunoglobulin mucin-3 (TIM-3),28 and T-cell immunoglobulin and ITIM domain (TIGIT).29 Targeting those pathways could be more effective than PD-1 blockade in FL; it is also possible that combination blockade could be synergistic. However, because only a small number of patients were classified as responders (n = 4), the number of biopsy samples available for analysis was also low. The associations found between patient response and biomarkers are only hypothesis generating, and their validity will need to be confirmed in subsequent studies.
Our results further strengthen the notion that the tumor microenvironment in FL plays an important role in the course of the disease. The IR-2 signature, comprising genes preferentially expressed in macrophages, dendritic cells, or both, was previously identified as a predictive marker of unfavorable prognosis in FL.16 In support of this, we found that higher expression of tumor-associated macrophage genes (TLR5, C3AR1, and FCGR1A) within IR-2 was associated with an increased risk of disease progression in patients treated with nivolumab. Thus, an immune-related gene signature may be predictive of responsiveness both to conventional therapy and to checkpoint blockade, providing a possible basis for trials combining PD-1/PD-L1 blockade with chemotherapy in FL; one such trial has already reported encouraging preliminary results.30 Additionally, we found that high serum levels of sIL-2Rα at baseline were associated with poor response to nivolumab, consistent with previous studies that demonstrated poor prognosis in patients with elevated sIL-2Rα/IL-2R who were treated with rituximab monotherapy or chemotherapy as first-line treatment of FL.31,32 Levels of sIL-2Rα are likely to reflect the state of the tumor microenvironment, on the basis of a positive correlation between the serum levels of sIL-2Rα and the number of CD68+ macrophages in FL.33
Overall, this trial demonstrates that single-agent PD-1 blockade is not effective in R/R FL. This negative finding may be important in several ways. First, it will help in the interpretation of results from trials that use PD-1 blockade in combination with other agents, by providing an estimate of the single-agent contribution of PD-1 blockade. Second, it may provide a note of caution in the design of combination studies that are not supported by biological hypotheses and help to guide the choice of rational combinations in patients with the appropriate biomarkers. Finally, if confirmed in future studies, the tumor characteristics found here to be associated with responsiveness to PD-1 blockade could form a basis for patient selection in future trials of checkpoint-based therapy.
The Bristol Myers Squibb policy on data sharing may be found at https://www.bms.com/researchers-and-partners/independent-research/data-sharing-request-process.html.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank all coinvestigators and the patients and families who participated in the trial, and Clarence K. Zhang, Bristol Myers Squibb (at the time of the study), for analysis of Edgeseq gene expression data. Editorial assistance was provided by Laura Yee, Namiko Abe, and Janice Zhou, of Caudex (New York, NY).
The biomarker studies involving sIL-2Rα were supported in part by funding from the Leukemia and Lymphoma Society. Editorial assistance was funded by Bristol Myers Squibb. P.A. gratefully acknowledges the support of the Harold and Virginia Lash Foundation and the Leukemia and Lymphoma Society. D.C. is funded by the National Institute for Health Research Biomedical Research Centre at the Royal Marsden and Institute of Cancer Research. S.M.A. acknowledges support from the Jaime Erin Follicular Research Consortium and the Leukemia and Lymphoma Society.
Authorship
Contribution: P.A. and S.M.A. designed the clinical study; S.J.R., S.S., and H.T. designed and contributed to the biomarker analyses; P.A., A.J., G.G., J.R., J.T., A.P., S.M.V., P.J., D.C., J.P.L., S.J.R., S.S., and S.M.A. collected data; P.A., S.J.R., P.M.-R., A.S., S.S., H.T., and S.M.A. analyzed and interpreted the data; S.J.R., P.M.-R., A.S., and H.T. performed statistical analysis; and all authors wrote, critically reviewed, and approved the manuscript before submission.
Conflict-of-interest disclosure: P.A. has consulted for Merck, Bristol Myers Squibb, Pfizer, Affimed, Adaptive, Infinity, ADC Therapeutics, and Celgene; received institutional research funding from Merck, Bristol Myers Squibb, Affimed, Adaptive, Roche, Tensha, Otsuka, Sigma Tau, Genentech, and IGM; and received honoraria from Merck and Bristol Myers Squibb. A.J. received an educational grant from Janssen; travel grants from Janssen, Celgene, AbbVie, and Roche; speaker fees from Janssen, Roche, AbbVie, Novartis, Amgen, Sanofi-Genzyme, and Celgene; and has consulted for Janssen, Roche, Gilead, AbbVie, Novartis, Amgen, and Sanofi-Genzyme. G.G. has consulted or advised for Autolus and Bioveloc ITA; served on a speakers’ bureau for Amgen; received research funding from Gilead; and received travel and accommodation from Roche, AbbVie, and Becton Dickinson. J.R. has received honoraria or advised for Takeda, Seattle Genetics, ADC Therapeutics, and Bristol Myers Squibb; received research funding from Takeda and ADC Therapeutics; and has stock ownership (spouse) in AstraZeneca and GlaxoSmithKline. J.T. holds stock or other ownership in Genmab, Corvus, Marker Therapeutics, and Bluebird Bio; has served in a consulting or advisory role with Kite, a Gilead Company, Celgene, Immune Design, and Celldex Therapeutics; has received research funding from Bristol Myers Squibb, Kite, a Gilead Company, Spectrum Pharmaceuticals, and Merck; and has received travel, accommodations, or expenses from Bristol Myers Squibb and Kite, a Gilead Company. A.P. has served on speakers’ bureaus for Roche, Celgene, Bristol Myers Squibb, Merck Sharp & Dohme, Servier, and Novartis. P.J. has received honoraria from Takeda, Bristol Myers Squibb, Novartis, Celgene, Kite Pharma, Genmab, and Incyte; has had a consulting or advisory role with Janssen Pharmaceuticals, Epizyme, and Boehringer Ingelheim; has received institutional research funding from Epizyme and Janssen Pharmaceuticals; has patents, royalties, or other intellectual property (combined use of Fc γ RIIb (CD32b) and CD20, specific antibodies, WO Patent, PCT/GB2011/051572; EU11760819.0); and has received travel, accommodation, and expenses from Zenyaku Kogyo. D.C. has received research funding from Amgen, Sanofi, Merrimack, AstraZeneca, Celgene, MedImmune, Bayer, 4SC, Clovis, Eli Lilly, Janssen, and Merck. J.P.L. has had a consulting or advisory role with Sutro, Gilead, Kite, AstraZeneca, Celgene, Roche/Genentech, ADC Therapeutics, Sandoz, Karyopharm, Miltenyi, Regeneron, and MEI Pharma. S.J.R. has received research funding from Bristol Myers Squibb, Affimed, KITE/Gilead, and Merck. P.M.-R. is an employee of and has equity ownership in Bristol Myers Squibb. A.S. is an employee of Bristol Myers Squibb. S.M.A. has received research funding from Bristol Myers Squibb, Merck, Seattle Genetics, Takeda, AI Therapeutics, Regeneron, Affimed, Trillium, and Pfizer. The remaining authors declare no competing financial interests.
Correspondence: Philippe Armand, Dana-Farber Cancer Institute, 450 Brookline Ave, Boston, MA, 02215; e-mail: philippe_armand@dfci.harvard.edu.