Key Points
Lethality of TM-null embryos is not a consequence of placental NLRP3 inflammasome activation.
EV-induced and platelet-mediated inflammasome activation reduces placental TM expression and proliferation, aiding fetal demise in PE.

Abstract
Excess platelet activation by extracellular vesicles (EVs) results in trophoblast inflammasome activation, interleukin 1β (IL-1β) activation, preeclampsia (PE), and partial embryonic lethality. Embryonic thrombomodulin (TM) deficiency, which causes embryonic lethality hallmarked by impaired trophoblast proliferation, has been linked with maternal platelet activation. We hypothesized that placental TM loss, platelet activation, and embryonic lethality are mechanistically linked to trophoblast inflammasome activation. Here, we uncover unidirectional interaction of placental inflammasome activation and reduced placental TM expression: although inflammasome inhibition did not rescue TM-null embryos from lethality, the inflammasome-dependent cytokine IL-1β reduced trophoblast TM expression and impaired pregnancy outcome. EVs, known to induce placental inflammasome activation, reduced trophoblast TM expression and proliferation. Trophoblast TM expression correlated negatively with IL-1β expression and positively with platelet numbers and trophoblast proliferation in human PE placentae, implying translational relevance. Soluble TM treatment or placental TM restoration ameliorated the EV-induced PE-like phenotype in mice, preventing placental thromboinflammation and embryonic death. The lethality of TM-null embryos is not a consequence of placental NLRP3 inflammasome activation. Conversely, EV-induced placental inflammasome activation reduces placental TM expression, promoting placental and embryonic demise. These data identify a new function of placental TM in PE and suggest that soluble TM limits thromboinflammatory pregnancy complications.
Introduction
Gestational vascular diseases, such as preeclampsia (PE), affect ∼5% to 7% of pregnancies and are a major cause of maternal, fetal, and neonatal morbidity and mortality.1 Gestational vascular diseases are closely associated with altered activation of the hemostatic system, but the underlying mechanisms remain poorly defined.2 Maternal and embryonic factors coordinately control aspects of placental hemostasis, including platelet activation.3-6 Placental thrombomodulin (TM) deficiency hinders normal placental development (eg, reduces trophoblast proliferation and increases trophoblast death) and results in embryonic lethality.5,7,8 Maternal platelet deficiency partially rescues TM-null embryos, establishing a crucial role of maternal platelet activation in the TM-dependent developmental phenotype.9 Intriguingly, excess platelet activation, for example, resulting from procoagulant extracellular vesicles (EVs), causes trophoblast inflammasome activation and results in a PE-like phenotype in mice.10 Decreased TM expression has been shown in PE patients and is possibly caused by an angiogenic imbalance.11 Accordingly, we initially speculated that loss of trophoblast TM expression unleashes platelet-dependent placental inflammasome activation, thus promoting embryonic lethality. However, our results imply a converse and unidirectional relationship of placental inflammasome activation and placental TM expression: inflammasome activation reduces placental TM expression and hence placental development and function.
Study design
Mice
Wild-type C57BL/6 mice were obtained from The Jackson Laboratory. TM-deficient and CMVCre mice have been published previously.8,12 TMhi-LoxP/LoxP mice were newly generated (supplemental Methods and supplemental Figure 7, available on the Blood Web site). Pregnant mice were injected with EVs as described earlier.10 Anakinra or soluble thrombomodulin (sTM; solulin) were injected intraperitoneally 30 minutes prior to each EV injection.10,13 All animal experiments were conducted following standards and procedures approved by the local Animal Care and Use Committee (Landesverwaltungsamt, Halle, Germany).
Human tissues
Human placenta samples from PE pregnancies and from normotensive control pregnancies (supplemental Table 1) were collected at the Universitätsfrauenklinik-Erlangen (Erlangen, Germany) in accordance with the guidelines and with the approval of the local Ethics Committee after obtaining informed patient consent.10
Generation of procoagulant EVs
EVs were prepared using previously described methods (see the supplemental Methods for details).10
Cell culture
Platelet-rich plasma (PRP) was prepared as previously described.10 Differentiated mouse trophoblast stem (mTS) cells were treated with interleukin 1β (IL-1β) or EV (final procoagulant activity, 7.5 nM thrombin equivalent) and PRP and, in some experiments, with anakinra or solulin along with EV and PRP for 24 hours in differentiation medium. Human trophoblast-like cells (JEG-3) were treated with EV (final procoagulant activity, 7.5 nM thrombin equivalent) and PRP for 24 hours.
Immunoblotting
Immunoblotting was conducted as previously described (see the supplemental Methods for details).10
Measurement of cell proliferation
Trophoblast proliferation was studied using a bromodeoxyuridine proliferation assay (in vitro) or Ki-67 immunohistochemistry (ex vivo).8
Statistical analysis
The data are summarized as the means ± standard error of the mean. Statistical analyses performed are delineated in the figure legends. Post hoc comparisons of analysis-of-variance (ANOVA) results were corrected with the Tukey method. The Kolmogorov-Smirnov test or D’Agostino-Pearson normality test was used to determine whether the distribution of the data were consistent with a Gaussian distribution. Prism 5 (www.graphpad.com) software was used for statistical analyses. Statistical significance was accepted at values of P < .05.
Results and discussion
To determine whether the lethality of TM-null embryos results from excess inflammasome activation, we treated pregnant TM+/− female mice mated with TM+/− male mice with the IL-1R antagonist anakinra (20 mg/kg). Although this dose efficiently prevented inflammasome-induced embryonic death,10 it did not improve the survival of TM-null embryos at day 10.5 postcoitus (Figure 1A). Similarly, a higher anakinra dose (40 mg/kg) or genetically superimposed Nlrp3 deficiency did not impact the lethality of TM-null embryos (Figure 1A). Hence, the lethality of TM-deficient embryos is independent of the NLRP3 inflammasome.
![Inflammasome activation induces loss of placental TM and trophoblast proliferation. (A) Stacked bar graph showing the frequency of embryos (red, TM+/+; green, TM−/−; blue, TM+/−). Anakinra treatment (either 20 or 40 mg/kg body weight, once daily starting on day 7.5 postcoitus) or genetic NLRP3 deficiency (Nlrp3−/−) do not rescue TM-null embryos (green bar) from lethality (n = 5 mothers per group). (B-C) Immunoblotting analysis (B, representative immunoblots; C, bar graph summarizing the results) showing a dose- and time-dependent (24 and 48 hours) reduction in TM expression upon treatment of differentiated mTS cells with exogenous IL-1β (n = 3 independent experiments). *P < .05 (relative to control; C, 24 hours); #P < .05 (relative to control; C, 48 hours), untreated controls, ANOVA (C). (D-G) Endothelial cell–derived EVs reduce TM expression in mice (D-E, n = 8 placentae from 3 different mothers), and anakinra treatment of EV-injected pregnant C57BL/6 mice restores TM expression. EVs reduce TM expression in differentiated mouse trophoblast cells (F-G, mTS; n = 3 independent experiments) or in human trophoblast-like cells (F-G, JEG-3; n = 3 independent experiments). Representative immunoblots (D,F). Bar graph summarizing the results (E). Line graph summarizing the results (G). *P < .05; Student t test (E); ANOVA (G). (H-J) Analysis of human placenta samples (n = 14 per group). Reduced TM expression (H) in PE patients compared with healthy pregnant controls (C), inverse correlation of TM expression and IL-β levels (I, PE patients, lavender; healthy pregnant controls [C], green), and positive correlation of TM expression and platelet counts (J). **P < .01; Student t test (H); Pearson correlation (I-J). (K-L) Ki-67 staining (brown, Ki-67+ nuclei; blue, hematoxylin nuclear counterstaining) in human placenta (K, representative images taken at ×40 magnification; L, dot plot with Pearson correlation; n = 14 per group) showing a positive association of TM expression and cell proliferation (PE patients [lavender]; healthy pregnant controls, C [green]; arrows, Ki-67+ nuclei). (M-O) Treatment of EV-injected pregnant C57BL/6 mice (M-N, n = 8 placentae from 3 different mothers per group) or differentiated mTS cells (O) with anakinra restores trophoblast cell proliferation (M, Ki-67 immunostaining, representative images taken at ×40 magnification; N, bar graph summarizing the results) and bromodeoxyuridine incorporation in mTS cells (O, bar graph summarizing the results). *P < .05 (relative to control [C]), #P < .05 (relative to EV); ANOVA (N,O). ANA, Anakinra; AU, arbitrary unit; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; ns, nonsignificant, χ2 test; UT, untreated control.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/7/10.1182_blood.2020005225/2/m_bloodbld2020005225f1.png?Expires=1766095677&Signature=3fx7JPWlt0vfqKBwHPwMFvaHcfAkqY5sfhLsrcPZ33vWzp6XVYmy0os5LNLVKqR-l1TkegPDk6~U8pDvxDCl1NfTdY15lXr1w8LwTf7up9--wkyZmMScRO~BO1Ie3joMOSrEdRhYWLA3lrv8QFtQ5kxB1IKbScv4ohDZx1IJSx1ooUH-04Cvz99QBfJO95K~TqR8uk9~G41cFbHGd~3BMsc4akgBUj8ktppS2apmEjPYhxbcNoCzPWKBpI09cU7W-sTJFgcyIe-KBefR~glRvSmhbj7stMmKPf0u5xP0YBT9L~N-2nmMpdc2sgpQrPxA5SDsq2tIsK4P8r5urv5oHQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Inflammasome activation induces loss of placental TM and trophoblast proliferation. (A) Stacked bar graph showing the frequency of embryos (red, TM+/+; green, TM−/−; blue, TM+/−). Anakinra treatment (either 20 or 40 mg/kg body weight, once daily starting on day 7.5 postcoitus) or genetic NLRP3 deficiency (Nlrp3−/−) do not rescue TM-null embryos (green bar) from lethality (n = 5 mothers per group). (B-C) Immunoblotting analysis (B, representative immunoblots; C, bar graph summarizing the results) showing a dose- and time-dependent (24 and 48 hours) reduction in TM expression upon treatment of differentiated mTS cells with exogenous IL-1β (n = 3 independent experiments). *P < .05 (relative to control; C, 24 hours); #P < .05 (relative to control; C, 48 hours), untreated controls, ANOVA (C). (D-G) Endothelial cell–derived EVs reduce TM expression in mice (D-E, n = 8 placentae from 3 different mothers), and anakinra treatment of EV-injected pregnant C57BL/6 mice restores TM expression. EVs reduce TM expression in differentiated mouse trophoblast cells (F-G, mTS; n = 3 independent experiments) or in human trophoblast-like cells (F-G, JEG-3; n = 3 independent experiments). Representative immunoblots (D,F). Bar graph summarizing the results (E). Line graph summarizing the results (G). *P < .05; Student t test (E); ANOVA (G). (H-J) Analysis of human placenta samples (n = 14 per group). Reduced TM expression (H) in PE patients compared with healthy pregnant controls (C), inverse correlation of TM expression and IL-β levels (I, PE patients, lavender; healthy pregnant controls [C], green), and positive correlation of TM expression and platelet counts (J). **P < .01; Student t test (H); Pearson correlation (I-J). (K-L) Ki-67 staining (brown, Ki-67+ nuclei; blue, hematoxylin nuclear counterstaining) in human placenta (K, representative images taken at ×40 magnification; L, dot plot with Pearson correlation; n = 14 per group) showing a positive association of TM expression and cell proliferation (PE patients [lavender]; healthy pregnant controls, C [green]; arrows, Ki-67+ nuclei). (M-O) Treatment of EV-injected pregnant C57BL/6 mice (M-N, n = 8 placentae from 3 different mothers per group) or differentiated mTS cells (O) with anakinra restores trophoblast cell proliferation (M, Ki-67 immunostaining, representative images taken at ×40 magnification; N, bar graph summarizing the results) and bromodeoxyuridine incorporation in mTS cells (O, bar graph summarizing the results). *P < .05 (relative to control [C]), #P < .05 (relative to EV); ANOVA (N,O). ANA, Anakinra; AU, arbitrary unit; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; ns, nonsignificant, χ2 test; UT, untreated control.
Inflammasome activation induces loss of placental TM and trophoblast proliferation. (A) Stacked bar graph showing the frequency of embryos (red, TM+/+; green, TM−/−; blue, TM+/−). Anakinra treatment (either 20 or 40 mg/kg body weight, once daily starting on day 7.5 postcoitus) or genetic NLRP3 deficiency (Nlrp3−/−) do not rescue TM-null embryos (green bar) from lethality (n = 5 mothers per group). (B-C) Immunoblotting analysis (B, representative immunoblots; C, bar graph summarizing the results) showing a dose- and time-dependent (24 and 48 hours) reduction in TM expression upon treatment of differentiated mTS cells with exogenous IL-1β (n = 3 independent experiments). *P < .05 (relative to control; C, 24 hours); #P < .05 (relative to control; C, 48 hours), untreated controls, ANOVA (C). (D-G) Endothelial cell–derived EVs reduce TM expression in mice (D-E, n = 8 placentae from 3 different mothers), and anakinra treatment of EV-injected pregnant C57BL/6 mice restores TM expression. EVs reduce TM expression in differentiated mouse trophoblast cells (F-G, mTS; n = 3 independent experiments) or in human trophoblast-like cells (F-G, JEG-3; n = 3 independent experiments). Representative immunoblots (D,F). Bar graph summarizing the results (E). Line graph summarizing the results (G). *P < .05; Student t test (E); ANOVA (G). (H-J) Analysis of human placenta samples (n = 14 per group). Reduced TM expression (H) in PE patients compared with healthy pregnant controls (C), inverse correlation of TM expression and IL-β levels (I, PE patients, lavender; healthy pregnant controls [C], green), and positive correlation of TM expression and platelet counts (J). **P < .01; Student t test (H); Pearson correlation (I-J). (K-L) Ki-67 staining (brown, Ki-67+ nuclei; blue, hematoxylin nuclear counterstaining) in human placenta (K, representative images taken at ×40 magnification; L, dot plot with Pearson correlation; n = 14 per group) showing a positive association of TM expression and cell proliferation (PE patients [lavender]; healthy pregnant controls, C [green]; arrows, Ki-67+ nuclei). (M-O) Treatment of EV-injected pregnant C57BL/6 mice (M-N, n = 8 placentae from 3 different mothers per group) or differentiated mTS cells (O) with anakinra restores trophoblast cell proliferation (M, Ki-67 immunostaining, representative images taken at ×40 magnification; N, bar graph summarizing the results) and bromodeoxyuridine incorporation in mTS cells (O, bar graph summarizing the results). *P < .05 (relative to control [C]), #P < .05 (relative to EV); ANOVA (N,O). ANA, Anakinra; AU, arbitrary unit; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; ns, nonsignificant, χ2 test; UT, untreated control.
We next hypothesized that trophoblast inflammasome activation and associated IL-1β release reduce trophoblast TM expression and that reduced trophoblast TM expression contributes to embryonic demise associated with placental inflammasome activation. Indeed, exposure of differentiated mTS cells to IL-1β reduced TM expression (messenger RNA and protein) and promoted TM shedding (Figure 1B-C; supplemental Figure 1). Furthermore, injection of endothelial cell–derived EVs into pregnant C57BL/6 mice at days 10.5 and 11.5 postcoitus, which causes placental inflammasome activation,10 reduced trophoblast TM expression (Figure 1D-E; supplemental Figure 2). Similarly, exposure of mTS cells or human placenta-derived JEG-3 (choriocarcinoma) cells to increasing numbers of EVs in the presence of platelets reduced TM expression in vitro (Figure 1F-G; supplemental Figure 1). Importantly, anakinra restored placental TM expression in EV-injected pregnant C57BL/6 mice, showing that inflammasome activation impairs placental TM expression (Figure 1D-E).
To scrutinize the translational relevance, we analyzed human placenta samples from PE patients and healthy controls. TM expression was reduced in trophoblast cells of PE placentae compared with control placentae whereas a reduction of TM expression was not detectable in embryonic endothelial cells (Figure 1H; supplemental Figure 2). Furthermore, TM expression correlated negatively with IL-1β expression and correlated positively with platelet counts in pregnant women without or with PE (Figure 1I-J).
Hence, although embryonic lethality of TM-deficient embryos is not a consequence of NLRP3 inflammasome activation, we speculated that the reverse is true: inflammasome activation, as observed in PE, reduces placental TM expression, which impairs placental development (eg, reduced trophoblast proliferation) and embryonic outcome.8 Indeed, TM expression in human PE placentae correlated positively with trophoblast proliferation (Figure 1K-L), another consequence of placental TM deficiency.8 In pregnant mice or in mTS cells, EVs reduced trophoblast proliferation (supplemental Figure 3), which was restored upon anakinra treatment (Figure 1M-O). Therefore, inflammasome-associated loss of TM promotes trophoblast growth arrest, reflecting the findings in TM-null placentae.8
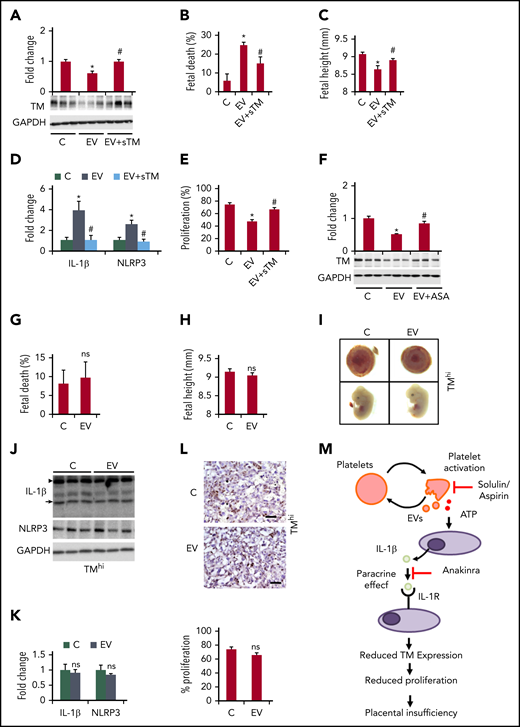
To scrutinize the functional relevance of reduced TM expression to the placental and embryonic defects associated with placental inflammasome activation, we aimed to restore TM levels. First, we used solulin, a modified version of sTM with increased stability,14 which markedly reduced EV-induced platelet activation (supplemental Figure 4). sTM prevented EV-induced and platelet-mediated inflammasome activation, as indicated by NLRP3 expression and IL-1β cleavage in mTS cells (supplemental Figure 5). Furthermore, upon solulin treatment, placental TM expression was maintained and EV-induced fetal death, intrauterine growth restriction, placental inflammasome activation, and reduced trophoblast proliferation were prevented (Figure 2A-E; supplemental Figure 6a-c). In vitro, solulin restored cell proliferation (supplemental Figure 6d). Aspirin treatment of EV-injected pregnant mice likewise maintained placental TM expression, indicating that platelet inhibition is sufficient to sustain TM expression (Figure 2F).

Restoring TM prevents EV-induced placental inflammasome activation and pregnancy failure. (A-E) Treatment of EV-injected pregnant C57BL/6 mice with solulin (1 mg/kg body weight) improves pregnancy outcomes. Bar graphs showing placental TM expression (A, bottom: representative immunoblots; GAPDH: loading control), fetal death (B), embryonic height (C), placental inflammasome activation, as reflected by IL-1β and NLRP3 (D, bar graph summarizing the results of immunoblot analyses), and placental cell proliferation (E, assessed by Ki-67 immunostaining, bar graph summarizing the results); n = 8 placentae from 3 different mothers; *P < .05 (relative to control, C), #P < .05 (relative to EV); ANOVA (A-E). (F) Aspirin treatment prevents the loss of placental TM expression (F, bar graph summarizing the results of immunoblot analyses; bottom, representative immunoblots; GAPDH, loading control). n = 8 placentae from 3 different mothers; *P < .05 (relative to control, C); #P < .05 (relative to EV); ANOVA. (G-L) Restoring placental TM expression prevents EV-induced pregnancy failure. Bar graphs showing fetal death (G) or embryonic height (H) and representative images showing the gross morphology of placentae (I, top) and embryos (I, bottom, n = 5 different mothers). Immunoblot images of IL-1β and NLRP3 (J, representative immunoblot; K, bar graph summarizing the results; n = 8 placentae from 3 different mothers). The arrow and arrowhead indicate the cleaved and pro forms of IL-1β, respectively. Expression of the cleaved form was quantified. Ki-67 immunostaining (L, top, representative images taken at ×40 magnification; bottom, bar graph summarizing results; n = 8 placentae from 3 different mothers). *P < .05; Student t test (G-H,K-L). (M) Proposed model summarizing results. EVs and platelet-induced inflammasome activation in trophoblast cells reduce trophoblast TM expression, reducing trophoblast proliferation and causing placental insufficiency. Note that loss of trophoblast TM expression is “downstream” of placental inflammasome activation and hence inflammasome inhibition does not rescue genetic TM deficiency. This model hence describes a unidirectional mechanism through which increased maternal platelet activation, as observed in PE, results in placental insufficiency and embryonic demise. Inhibiting this thromboinflammatory mechanism by solulin or anakinra can restore placental TM levels. ASA, acetylsalicylic acid (aspirin); ATP, adenosine triphosphate.
Restoring TM prevents EV-induced placental inflammasome activation and pregnancy failure. (A-E) Treatment of EV-injected pregnant C57BL/6 mice with solulin (1 mg/kg body weight) improves pregnancy outcomes. Bar graphs showing placental TM expression (A, bottom: representative immunoblots; GAPDH: loading control), fetal death (B), embryonic height (C), placental inflammasome activation, as reflected by IL-1β and NLRP3 (D, bar graph summarizing the results of immunoblot analyses), and placental cell proliferation (E, assessed by Ki-67 immunostaining, bar graph summarizing the results); n = 8 placentae from 3 different mothers; *P < .05 (relative to control, C), #P < .05 (relative to EV); ANOVA (A-E). (F) Aspirin treatment prevents the loss of placental TM expression (F, bar graph summarizing the results of immunoblot analyses; bottom, representative immunoblots; GAPDH, loading control). n = 8 placentae from 3 different mothers; *P < .05 (relative to control, C); #P < .05 (relative to EV); ANOVA. (G-L) Restoring placental TM expression prevents EV-induced pregnancy failure. Bar graphs showing fetal death (G) or embryonic height (H) and representative images showing the gross morphology of placentae (I, top) and embryos (I, bottom, n = 5 different mothers). Immunoblot images of IL-1β and NLRP3 (J, representative immunoblot; K, bar graph summarizing the results; n = 8 placentae from 3 different mothers). The arrow and arrowhead indicate the cleaved and pro forms of IL-1β, respectively. Expression of the cleaved form was quantified. Ki-67 immunostaining (L, top, representative images taken at ×40 magnification; bottom, bar graph summarizing results; n = 8 placentae from 3 different mothers). *P < .05; Student t test (G-H,K-L). (M) Proposed model summarizing results. EVs and platelet-induced inflammasome activation in trophoblast cells reduce trophoblast TM expression, reducing trophoblast proliferation and causing placental insufficiency. Note that loss of trophoblast TM expression is “downstream” of placental inflammasome activation and hence inflammasome inhibition does not rescue genetic TM deficiency. This model hence describes a unidirectional mechanism through which increased maternal platelet activation, as observed in PE, results in placental insufficiency and embryonic demise. Inhibiting this thromboinflammatory mechanism by solulin or anakinra can restore placental TM levels. ASA, acetylsalicylic acid (aspirin); ATP, adenosine triphosphate.
To determine whether restoring TM expression in embryonic placentae is sufficient to rescue EV-induced and inflammasome-mediated embryonic demise, we genetically restored TM expression in the placenta. We generated mice with conditional (Cre-dependent) TM expression (TMhi-LoxP/LoxP mice; supplemental Figure 7). Male TMhi-LoxP/LoxP mice were mated with female mice homozygous for X-linked Cre recombinase (CMVCre mice; homozygous mice were obtained by inbreeding and genotyping the progeny). This strategy resulted in Cre-mediated, constitutive TM expression only within embryonic tissues, including trophoblast cells (supplemental Figure 7). These pregnant female mice were injected with EVs at days 10.5 and 11.5 postcoitus. Genetically maintained placental TM expression protected against EV-induced fetal death, intrauterine growth restriction, and placental inflammasome activation, and restored trophoblast proliferation (Figure 2G-L). Taken together, these findings show that restoration of placental TM levels in pregnant mice, either upon systemically inhibiting platelet activation (using solulin or aspirin) or locally within the placenta, prevents EV-induced placental inflammasome activation, placental dysfunction, and embryonic demise.
These results establish a unidirectional relation of inflammasome activation and reduced trophoblast TM expression, which contributes to the placental defects and embryonic demise observed in PE (Figure 2M). The impaired survival and growth of embryos with partially reduced TM expression in the placenta secondary to EV-induced maternal PE is in agreement with the impaired survival and development of embryos with genetically superimposed partial loss of TM function and additional maternal risk factors.15 Restoring TM levels may be a new and, as solulin cannot cross the placental barrier, safe therapy for placental defects associated with excess platelet activation and placental inflammasome activation, as observed in PE.10,16-18 However, our data do not support the converse hypothesis that the placental defects observed in TM-null embryos are mechanistically linked to NLRP3 inflammasome activation. Whether platelet-dependent placental inflammasome activation may be a contributing factor to pregnancy complications in less severe cases of thrombophilia, such as those observed in pregnancies of homozygous factor V Leiden carriers,15 remains to be established.
For original data, please e-mail the corresponding authors, Shrey Kohli and Berend Isermann, at shrey.kohli@medizin.uni-leipzig.de and berend.isermann@medizin.uni-leipzig.de, respectively.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Kathrin Deneser, Julia Judin, René Rudat, and Rumiya Makarova for excellent technical support. The authors also thank Satish Ranjan for support in some animal experiments.
This work was supported by Deutsche Forschungsgemeinschaft grants KO-5736/1-1 (S.K.), 361210922/GRK2408 (B.I.), IS-67/4-3 (B.I.), and IS-67/16-1 (B.I.).
Authorship
Contribution: S.K. designed, performed, and interpreted the results of the in vivo, in vitro, and ex vivo experiments and prepared the manuscript; K.K.S., P.M., F.L., D.G., A.G., R.R., and A.E. supported the mouse experiments and ex vivo analysis; H.H. and M.R. provided human placenta samples; and B.I. designed and interpreted the experimental work and prepared the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Shrey Kohli, Institute of Laboratory Medicine, Clinical Chemistry, and Molecular Diagnostics, Leipzig University, Liebigstr 27A, 04103 Leipzig, Germany; e-mail: shrey.kohli@medizin.uni-leipzig.de; and Berend Isermann, Institute of Laboratory Medicine, Clinical Chemistry, and Molecular Diagnostics, Leipzig University, Liebigstr 27A, 04103 Leipzig, Germany; e-mail: berend.isermann@medizin.uni-leipzig.de.


![Inflammasome activation induces loss of placental TM and trophoblast proliferation. (A) Stacked bar graph showing the frequency of embryos (red, TM+/+; green, TM−/−; blue, TM+/−). Anakinra treatment (either 20 or 40 mg/kg body weight, once daily starting on day 7.5 postcoitus) or genetic NLRP3 deficiency (Nlrp3−/−) do not rescue TM-null embryos (green bar) from lethality (n = 5 mothers per group). (B-C) Immunoblotting analysis (B, representative immunoblots; C, bar graph summarizing the results) showing a dose- and time-dependent (24 and 48 hours) reduction in TM expression upon treatment of differentiated mTS cells with exogenous IL-1β (n = 3 independent experiments). *P < .05 (relative to control; C, 24 hours); #P < .05 (relative to control; C, 48 hours), untreated controls, ANOVA (C). (D-G) Endothelial cell–derived EVs reduce TM expression in mice (D-E, n = 8 placentae from 3 different mothers), and anakinra treatment of EV-injected pregnant C57BL/6 mice restores TM expression. EVs reduce TM expression in differentiated mouse trophoblast cells (F-G, mTS; n = 3 independent experiments) or in human trophoblast-like cells (F-G, JEG-3; n = 3 independent experiments). Representative immunoblots (D,F). Bar graph summarizing the results (E). Line graph summarizing the results (G). *P < .05; Student t test (E); ANOVA (G). (H-J) Analysis of human placenta samples (n = 14 per group). Reduced TM expression (H) in PE patients compared with healthy pregnant controls (C), inverse correlation of TM expression and IL-β levels (I, PE patients, lavender; healthy pregnant controls [C], green), and positive correlation of TM expression and platelet counts (J). **P < .01; Student t test (H); Pearson correlation (I-J). (K-L) Ki-67 staining (brown, Ki-67+ nuclei; blue, hematoxylin nuclear counterstaining) in human placenta (K, representative images taken at ×40 magnification; L, dot plot with Pearson correlation; n = 14 per group) showing a positive association of TM expression and cell proliferation (PE patients [lavender]; healthy pregnant controls, C [green]; arrows, Ki-67+ nuclei). (M-O) Treatment of EV-injected pregnant C57BL/6 mice (M-N, n = 8 placentae from 3 different mothers per group) or differentiated mTS cells (O) with anakinra restores trophoblast cell proliferation (M, Ki-67 immunostaining, representative images taken at ×40 magnification; N, bar graph summarizing the results) and bromodeoxyuridine incorporation in mTS cells (O, bar graph summarizing the results). *P < .05 (relative to control [C]), #P < .05 (relative to EV); ANOVA (N,O). ANA, Anakinra; AU, arbitrary unit; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; ns, nonsignificant, χ2 test; UT, untreated control.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/7/10.1182_blood.2020005225/2/m_bloodbld2020005225f1.png?Expires=1766095678&Signature=ZBHs6nHoXyAXKPAg6vws80u4RJZVxfvnKQ0quQBlndFVfbs7zWDpkh-R4QszdF8kgZEas7sh3lGloO0qbCiXzqxkt-opayW24nYTcE4wmlqoowLaPKSfS5FBWd1p6EXgf7mqeAutqercQA5-FSn2olu0ps~2AiqArK01KqqC6yLxVjx19H1beMENVGqFzgkGH5KH6GZdS1rnIcNPsoVGGBHmpgW8uW2ULDxubSMbi4tpKQutWYhbOjpIzLr6DtnR-uMLfmKFtAhgXIHNXPXbsGErzEv3YjmPxmc-ofAkWWSUcPZVbadiyBvfuWDQFyfRjdBWCs-ENLZiwny6nbEXuw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)