Key Points
Intermittent blockade of IFN-γ signaling by JAK inhibitors is inadequate to prevent development of experimental HLH.
Addition of ruxolitinib to anti-IFN-γ worsens survival of experimental animals with HLH from toxicity of JAK2 inhibition.

Abstract
Hemophagocytic lymphohistiocytosis (HLH) is an inflammatory disorder in which numerous cytokines are elevated, though interferon-γ (IFN-γ) is central to disease pathogenesis and a key therapeutic target. Experimental and early clinical reports have shown that ruxolitinib, a small molecule inhibitor of Janus kinases (JAKs), which are essential for cytokine signaling, may be therapeutic in HLH. In contrast, we found that intermittently administered ruxolitinib at various dose levels failed to prevent HLH development or treat established murine HLH. High doses of ruxolitinib blocked IFN-γ signaling only transiently after administration, consistent with human pharmacokinetics, and only continuously administered drug could prevent HLH development or treat established HLH. Continuously administered ruxolitinib was therapeutic in only a narrow dose range and intermittently dosed ruxolitinib worsened survival and decreased bone marrow cellularity of animals concurrently treated with anti-IFN-γ antibody, indicating a narrow therapeutic window and potential toxicity. Because JAK2 is essential for hematopoietic cytokine signaling, we also tested a JAK1-selective inhibitor and observed therapeutic benefit without apparent toxicity, though it did not improve survival when combined with anti-IFN-γ. We conclude that continuous blockade of IFN-γ signaling is necessary for optimal control of HLH and that JAK2 inhibition may be toxic in this disorder.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a hyperinflammatory disorder in which numerous cytokines are elevated, exemplifying the concept of a “cytokine storm.”1 Primary HLH has been modeled in mice by multiple groups, demonstrating that interferon-γ (IFN-γ) plays a key role in driving disease pathology and leading to the clinical development of emapalumab, a therapeutic anti-IFN-γ antibody.2-6 The centrality of IFN-γ in HLH has also led to interest in inhibiting janus kinases (JAKs), which are essential for signaling by IFN-γ and other cytokines. Experimental studies and early clinical reports have indicated that ruxolitinib may have utility in experimental and clinical HLH, though mostly in secondary HLH.7-11 Because ruxolitinib inhibits both JAK1 and JAK2, it causes broad inhibition of cytokine signaling, which could be advantageous in HLH.12 We performed studies to better understand how JAK inhibitors may be applied in a rigorous model of primary HLH. We unexpectedly found that intermittent blockade of IFN-γ signaling substantially hindered therapeutic effects of ruxolitinib, continuously administered ruxolitinib had a narrow therapeutic dose range, and that inhibition of JAK2 (but not JAK1) was associated with notable toxicity. Thus, these data suggest cautious appraisal of the clinical potential of broadly acting small molecule JAK inhibitors in the context of HLH.
Methods
Reagents
Ruxolitinib was obtained from the CCHMC pharmacy or Selleckchem and prepared/administered exactly as described.7,8 Alternatively, ruxolitinib was given intraperitoneally in some animals or in drinking water (also supplemented with 0.1 mg/mL Trappsol and 0.3 mg/mL saccharin). AZD4205 (25 mg/kg per dose, gavage twice daily) was obtained from Chemietek. Anti-IFN-γ (XMG1.2) was given every 3 to 4 days after lymphocytic choriomeningitis virus (LCMV) infection intraperitoneally at 40 mg/kg per dose.
Mice/treatments
Animal experiments were performed with IACUC approval. HLH model and scoring system were as previously described.13 Briefly, mice were infected with LCMV (strain WE, 200 pfu) intraperitoneally and monitored daily. Death occurred spontaneously or from euthanasia triggered by >30% weight loss or moribund status, per a predefined scoring system.13 IFN-γ was infused via osmotic pump as described previously.14
Results and discussion
Continuous blockade of IFN-γ signaling is necessary for control of HLH
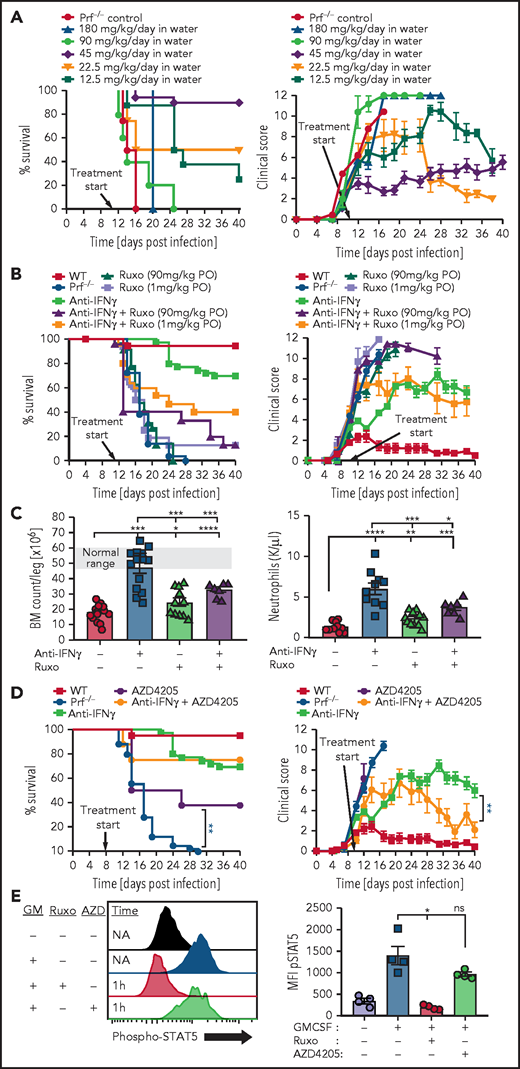
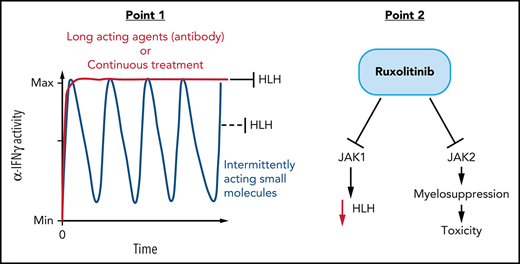
To study ruxolitinib’s therapeutic potential in HLH, we treated mice twice daily as previously reported with either low-dose (1 mg/kg per dose) or high-dose (90 mg/kg per dose) ruxolitinib.7,8 Using our well-established model of primary HLH consisting of perforin-deficient (prf−/−) mice challenged with LCMV,2 we found that ruxolitinib did not display substantial therapeutic benefit (Figure 1A), in contrast to prior reports.7,8 We tested ruxolitinib across multiple experiments in almost 40 animals, varying dose and routes of administration with consistent results. Importantly, to mimic prior reports, we administered ruxolitinib in a preemptive fashion before onset of clinically detectable HLH (Figure 1A2,13), though this does not mimic clinical application in ill patients.13 although differences between HLH models used by us and different groups (eg, more vs less aggressive LCMV strains, differences in facility microbiome) may explain some differences with prior reports, we hypothesized that this limited benefit may also be related to ruxolitinib’s short half-life. We directly tested this hypothesis by measuring IFN-γ signaling at various times after ruxolitinib administration to animals and compared it with anti-IFN-γ antibody treatment. We found that although antibody blocked STAT1 phosphorylation in response to IFN-γ for an extended time in vivo, high-dose ruxolitinib was able to fully block STAT1 phosphorylation for only a few hours (Figure 1B). This result is consistent with ruxolitinib’s pharmacokinetics in humans, where levels sufficient to fully block IFN-γ signaling are only achieved for 2 to 6 hours after a dose.15-17 Next, we directly tested the differential impact of intermittent vs continuous IFN-γ blockade by comparing conventionally (intermittently) dosed ruxolitinib and anti-IFN-γ antibody in animals infused with IFN-γ. We previously showed that prolonged infusion of IFN-γ alone (without LCMV infection) recreates features of HLH, including splenomegaly, anemia, and hemophagocytosis.14 We found that high-dose ruxolitinib only partially prevented development of splenomegaly and was unable to block development of anemia, whereas anti-IFN-γ antibody was fully effective (Figure 1C). Finally, we administered ruxolitinib continuously by adding it to the drinking water of LCMV-infected prf−/− mice. In contrast to conventional (intermittent) administration, we found that continuous administration prevented the development of HLH and provided extended blockade of IFN-γ signaling (Figure 1D-E). Taken together, we conclude that continuous (instead of intermittent) blockade of IFN-γ signaling is necessary for optimal control of HLH. This finding contrast with JAK inhibition for the treatment of myeloproliferative neoplasms, where intermittent blockade of JAK signaling is sufficient to halt abnormal cellular proliferation. This insight also suggests that alternative schedules of ruxolitinib administration or other JAK inhibitors with longer half-lives may be more clinically effective.
![Continuous blockade of IFN-γ signaling is necessary for prevention of HLH and IFN-γ mediated pathologies. (A) Survival and global clinical score of perforin-deficient (prf−/−) mice after LCMV infection and ruxolitinib treatment. All agents were started 5 days after infection, before onset of HLH, as preemptive treatments (as indicated by clinical score13). Two dose levels of ruxolitinib (1 or 90 mg/kg per dose, oral gavage [PO] twice daily, as previously reported7,8) and 2 routes of administration (oral or intraperitoneal [IP]) were tested. For comparison, wild-type (WT) mice and prf−/− mice given control treatments (carrier gavage) are shown. (B) Kinetic assessment of IFN-γ signaling blockade in vivo by ruxolitinib. Mice were treated with ruxolitinib (90 mg/kg, IP) or anti-IFN-γ antibody (40 mg/kg XMG1.2) at the indicated times before injection with IFN-γ (300 ng, IP). Twenty minutes later, animals were euthanized and peritoneal macrophages were harvested via peritoneal lavage with 1% paraformaldehyde. After methanol permeabilization, phospho-STAT1 was measured in F4/80+ cells by flow cytometry. (C) Development of splenomegaly and anemia after IFN-γ infusion. Uninfected WT mice were infused with IFN-γ (or phosphate-buffered saline) via osmotic pumps (as described previously14) and spleen weights and blood hemoglobin levels were assessed 6 days later. Mice were simultaneously treated with saline, ruxolitinib (90 mg/kg, twice daily), or anti-IFN-γ antibody as indicated. (D) Survival and clinical score after LCMV infection of prf−/− mice treated with ruxolitinib administered continuously in the drinking water (0.5 mg/mL, delivering 45 mg/kg per day). (E) Assessment of IFN-γ signaling blockade in vivo by continuously administered ruxolitinib. Mice received ruxolitinib in the drinking water (0.5 mg/mL) for 12 hours and were assessed as described in B. N = 5-39 mice/group (39 mice total treated with ruxolitinib in panel A) combined from 3 to 12 independent experiments. *P < .05, **P < .01, ***P < .001.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/12/10.1182_blood.2020007930/4/m_bloodbld2020007930r2f1.png?Expires=1767791124&Signature=qyNXDuxGiRmcyKY3W0jVcXWgIn3bmNIuPSb6SlGKzTMpCPY-Tcrs5PlddFNg-zrbcs6TbHnjyu7q8gnY0-NUMr3C5vB8O3Li8AR~Ix4aAtoSzCvNGEfACkVkQ5uXlIGrgqeqXXFx7CuFe~Rl9RgoVTjjra3RrzDgW3nxCoOk9xg9pP1x3-tXQTueIAxNyPVA7Cra9-s1KbbfyLw68QvwToU2rDjY4lG0jae1JEb~2CQoOqHXiA893kiqNIRmW-Glozf4gLWV8Oc~xOoDoFMCcQi1o~IYbSV91BocKp5KM82gMn3ITwr1qwsr5otIF3FQrkrR85x6U5MINOmFblIH7Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Continuous blockade of IFN-γ signaling is necessary for prevention of HLH and IFN-γ mediated pathologies. (A) Survival and global clinical score of perforin-deficient (prf−/−) mice after LCMV infection and ruxolitinib treatment. All agents were started 5 days after infection, before onset of HLH, as preemptive treatments (as indicated by clinical score13). Two dose levels of ruxolitinib (1 or 90 mg/kg per dose, oral gavage [PO] twice daily, as previously reported7,8) and 2 routes of administration (oral or intraperitoneal [IP]) were tested. For comparison, wild-type (WT) mice and prf−/− mice given control treatments (carrier gavage) are shown. (B) Kinetic assessment of IFN-γ signaling blockade in vivo by ruxolitinib. Mice were treated with ruxolitinib (90 mg/kg, IP) or anti-IFN-γ antibody (40 mg/kg XMG1.2) at the indicated times before injection with IFN-γ (300 ng, IP). Twenty minutes later, animals were euthanized and peritoneal macrophages were harvested via peritoneal lavage with 1% paraformaldehyde. After methanol permeabilization, phospho-STAT1 was measured in F4/80+ cells by flow cytometry. (C) Development of splenomegaly and anemia after IFN-γ infusion. Uninfected WT mice were infused with IFN-γ (or phosphate-buffered saline) via osmotic pumps (as described previously14) and spleen weights and blood hemoglobin levels were assessed 6 days later. Mice were simultaneously treated with saline, ruxolitinib (90 mg/kg, twice daily), or anti-IFN-γ antibody as indicated. (D) Survival and clinical score after LCMV infection of prf−/− mice treated with ruxolitinib administered continuously in the drinking water (0.5 mg/mL, delivering 45 mg/kg per day). (E) Assessment of IFN-γ signaling blockade in vivo by continuously administered ruxolitinib. Mice received ruxolitinib in the drinking water (0.5 mg/mL) for 12 hours and were assessed as described in B. N = 5-39 mice/group (39 mice total treated with ruxolitinib in panel A) combined from 3 to 12 independent experiments. *P < .05, **P < .01, ***P < .001.
Continuous blockade of IFN-γ signaling is necessary for prevention of HLH and IFN-γ mediated pathologies. (A) Survival and global clinical score of perforin-deficient (prf−/−) mice after LCMV infection and ruxolitinib treatment. All agents were started 5 days after infection, before onset of HLH, as preemptive treatments (as indicated by clinical score13). Two dose levels of ruxolitinib (1 or 90 mg/kg per dose, oral gavage [PO] twice daily, as previously reported7,8) and 2 routes of administration (oral or intraperitoneal [IP]) were tested. For comparison, wild-type (WT) mice and prf−/− mice given control treatments (carrier gavage) are shown. (B) Kinetic assessment of IFN-γ signaling blockade in vivo by ruxolitinib. Mice were treated with ruxolitinib (90 mg/kg, IP) or anti-IFN-γ antibody (40 mg/kg XMG1.2) at the indicated times before injection with IFN-γ (300 ng, IP). Twenty minutes later, animals were euthanized and peritoneal macrophages were harvested via peritoneal lavage with 1% paraformaldehyde. After methanol permeabilization, phospho-STAT1 was measured in F4/80+ cells by flow cytometry. (C) Development of splenomegaly and anemia after IFN-γ infusion. Uninfected WT mice were infused with IFN-γ (or phosphate-buffered saline) via osmotic pumps (as described previously14) and spleen weights and blood hemoglobin levels were assessed 6 days later. Mice were simultaneously treated with saline, ruxolitinib (90 mg/kg, twice daily), or anti-IFN-γ antibody as indicated. (D) Survival and clinical score after LCMV infection of prf−/− mice treated with ruxolitinib administered continuously in the drinking water (0.5 mg/mL, delivering 45 mg/kg per day). (E) Assessment of IFN-γ signaling blockade in vivo by continuously administered ruxolitinib. Mice received ruxolitinib in the drinking water (0.5 mg/mL) for 12 hours and were assessed as described in B. N = 5-39 mice/group (39 mice total treated with ruxolitinib in panel A) combined from 3 to 12 independent experiments. *P < .05, **P < .01, ***P < .001.
Inhibition of JAK2, but not JAK1, is toxic during experimental HLH
To study ruxolitinib in a therapeutic context in addition to a preemptive one, we also treated animals starting on day 11 after LCMV infection, which is after the onset of clinical disease but before mortality is observed. We tested various concentrations of continuously administered ruxolitinib in the animals’ drinking water. We found that the same dose that effectively prevented HLH development (∼45 mg/kg per day) also treated established HLH, but that all doses tested above or below that amount were ineffective (Figure 2A). We also tested high or low doses of ruxolitinib administered conventionally (twice daily via oral gavage) as either monotherapy or in combination with anti-IFN-γ antibody. We did not observe therapeutic benefit as monotherapy and, surprisingly, we found that ruxolitinib decreased survival and worsened clinical scores of animals treated concurrently with anti-IFN-γ, which would ordinarily rescue most animals (Figure 2B).13
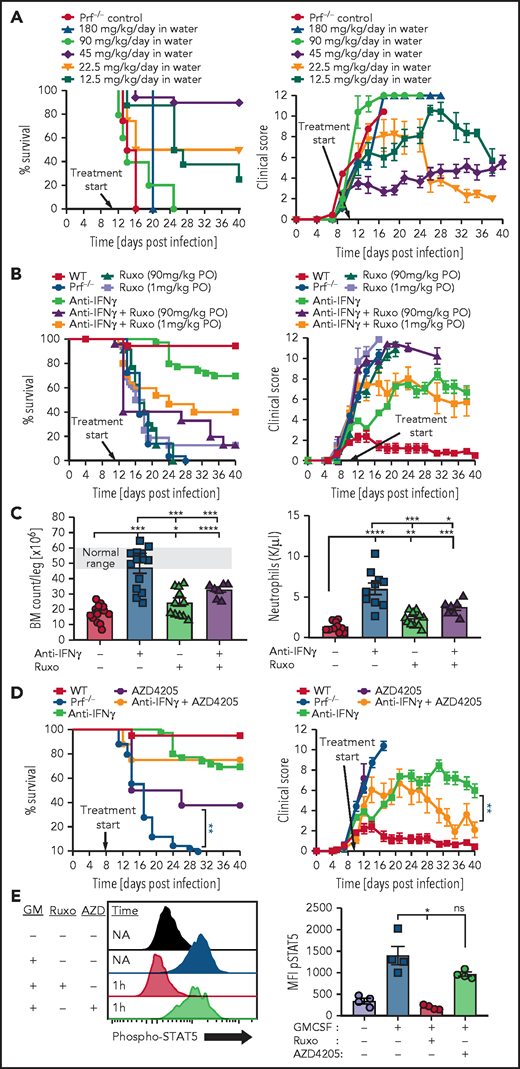
Ruxolitinib displays a narrow therapeutic window when treating established HLH and inhibition of JAK2, but not JAK1, is potentially toxic during HLH. (A) Survival and global clinical score of prf−/− mice treated after onset of HLH, on day 11, with continuously administered ruxolitinib in the drinking water at various doses. (B) Survival and global clinical score of prf−/− mice with established HLH treated with ruxolitinib, anti-IFN-γ, or combinations thereof. Anti-IFN-γ antibody was administered intraperitoneally (XMG1.2, 40 mg/kg, every 3-4 days) and ruxolitinib was given via oral gavage twice daily at the indicated doses. (C) Impact of ruxolitinib on marrow and peripheral blood cellularity. Bone marrow and peripheral blood neutrophil counts were assessed 16 days after infection, with the indicated treatments starting on day 6 (ruxolitinib was administered at 90 mg/kg per dose orally twice daily). (D) Survival and global clinical score of prf−/− mice after LCMV infection treated with the JAK1-selective inhibitor AZD4205, with or without anti-IFN-γ antibody. AZD4205 was administered as 25 mg/kg twice daily via oral gavage. (E) Assessment of JAK2 blockade in vivo by ruxolitinib and AZD4205. Mice were treated with ruxolitinib (90 mg/kg, IP) or AZD4205 (25 mg/kg, IP) and injected with granulocyte macrophage colony-stimulating factor (200 ng, IP) 1 hour later. Twenty minutes later, animals were euthanized and peritoneal macrophages were harvested via peritoneal lavage with 1% paraformaldehyde. After methanol permeabilization, phospho-STAT5 was measured in F4/80+ cells by flow cytometry. N = 8-37 mice/group (29 mice total treated with high- or low-dose ruxolitinib in panel A), combined from 2 to 12 independent experiments. *P < .05, **P < .01, ***P < .001.
Ruxolitinib displays a narrow therapeutic window when treating established HLH and inhibition of JAK2, but not JAK1, is potentially toxic during HLH. (A) Survival and global clinical score of prf−/− mice treated after onset of HLH, on day 11, with continuously administered ruxolitinib in the drinking water at various doses. (B) Survival and global clinical score of prf−/− mice with established HLH treated with ruxolitinib, anti-IFN-γ, or combinations thereof. Anti-IFN-γ antibody was administered intraperitoneally (XMG1.2, 40 mg/kg, every 3-4 days) and ruxolitinib was given via oral gavage twice daily at the indicated doses. (C) Impact of ruxolitinib on marrow and peripheral blood cellularity. Bone marrow and peripheral blood neutrophil counts were assessed 16 days after infection, with the indicated treatments starting on day 6 (ruxolitinib was administered at 90 mg/kg per dose orally twice daily). (D) Survival and global clinical score of prf−/− mice after LCMV infection treated with the JAK1-selective inhibitor AZD4205, with or without anti-IFN-γ antibody. AZD4205 was administered as 25 mg/kg twice daily via oral gavage. (E) Assessment of JAK2 blockade in vivo by ruxolitinib and AZD4205. Mice were treated with ruxolitinib (90 mg/kg, IP) or AZD4205 (25 mg/kg, IP) and injected with granulocyte macrophage colony-stimulating factor (200 ng, IP) 1 hour later. Twenty minutes later, animals were euthanized and peritoneal macrophages were harvested via peritoneal lavage with 1% paraformaldehyde. After methanol permeabilization, phospho-STAT5 was measured in F4/80+ cells by flow cytometry. N = 8-37 mice/group (29 mice total treated with high- or low-dose ruxolitinib in panel A), combined from 2 to 12 independent experiments. *P < .05, **P < .01, ***P < .001.
The narrow therapeutic window we observed and the negative impact of ruxolitinib on anti-IFN-γ based therapy suggested that ruxolitinib may be exerting both therapeutic and toxic effects in this model of established HLH. Notably, a prior report testing ruxolitinib in experimental HLH also noted drug-related toxicity, but these effects were not well described.8 We hypothesized that this apparent toxicity was due to myelosuppression by ruxolitinib because this is a known adverse effect of this agent18,19 and because it inhibits JAK2, which is essential for signaling of multiple cytokines that promote hematopoiesis.20 To test this idea, we examined bone marrow cellularity in prf−/− mice after initiating therapies on day 6 after infection and found that although anti-IFN-γ antibody treatment substantially improved marrow cellularity, cotreatment with ruxolitinib prevented this rescue (Figure 2C). We observed a similar pattern with peripheral blood neutrophil counts (Figure 2C). If the toxicity we observed from ruxolitinib was due to its effects on JAK2, then one would predict that a JAK1 selective inhibitor would have anti-HLH activity but would not worsen outcomes when combined with anti-IFN-γ antibody. Indeed, when we tested a JAK1 selective inhibitor (AZD4205),21 we observed a significant improvement in survival when used as a monotherapy for murine HLH, and we did not find evidence of toxicity when combined with anti-IFN-γ (Figure 2D). We confirmed that AZD4205 does not inhibit JAK2 at the therapeutic doses tested by examining JAK2-mediated STAT5 phosphorylation in peritoneal macrophages in response to GM-CSF. Although ruxolitinib completely inhibited STAT5 phosphorylation, AZD4205 had little effect (Figure 2E). In addition to demonstrating the potential toxicity of JAK2 inhibition in HLH, this is the first demonstration of the therapeutic benefit of a JAK1-selective inhibitor for HLH.
Overall, our data demonstrate that the therapeutic utility of JAK inhibitors for experimental HLH may be limited by only intermittently inhibiting IFN-γ signaling with conventional administration schedules. Optimal application of this class of drugs for HLH may require careful attention to drugs levels sufficient to inhibit IFN-γ signaling and potentially novel dosing strategies. Furthermore, our studies indicate that ruxolitinib may exert toxic effects from JAK2 inhibition/myelosuppression. Although a prior report suggested that ruxolitinib exerts anti-HLH effects via broad inhibition of cytokine signaling,12 our results indicate that this effect is more complex than previously thought, perhaps with both beneficial and harmful aspects. As the number of agents that may be potentially applied for the treatment of HLH expand, additional studies of novel combinations (such as reported recently22) and novel dosing regimens are increasingly needed. Although our data highlight issues not apparent in prior reports, they are consistent with ruxolitinib displaying some clinical activity as reported, but suggest that results are likely to be mixed in primary HLH patients, and provide additional reasons for caution when considering use of this agent outside of controlled clinical trials.
Acknowledgments
The authors thank Liam’s Lighthouse Foundation and the Neumann family foundation for support of these studies.
Authorship
Contribution: V.C. and M.B.J. planned experiments, analyzed data, and wrote the manuscript; and V.C., N.L., M.T., and N.C. planned and performed experiments.
Conflict-of-interest disclosure: M.B.J. has had a consulting or advisory role for Sobi/Novimmune, Bristol-Myers Squibb, and Alexion. The remaining authors declare no competing financial interests.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

![Continuous blockade of IFN-γ signaling is necessary for prevention of HLH and IFN-γ mediated pathologies. (A) Survival and global clinical score of perforin-deficient (prf−/−) mice after LCMV infection and ruxolitinib treatment. All agents were started 5 days after infection, before onset of HLH, as preemptive treatments (as indicated by clinical score13). Two dose levels of ruxolitinib (1 or 90 mg/kg per dose, oral gavage [PO] twice daily, as previously reported7,8) and 2 routes of administration (oral or intraperitoneal [IP]) were tested. For comparison, wild-type (WT) mice and prf−/− mice given control treatments (carrier gavage) are shown. (B) Kinetic assessment of IFN-γ signaling blockade in vivo by ruxolitinib. Mice were treated with ruxolitinib (90 mg/kg, IP) or anti-IFN-γ antibody (40 mg/kg XMG1.2) at the indicated times before injection with IFN-γ (300 ng, IP). Twenty minutes later, animals were euthanized and peritoneal macrophages were harvested via peritoneal lavage with 1% paraformaldehyde. After methanol permeabilization, phospho-STAT1 was measured in F4/80+ cells by flow cytometry. (C) Development of splenomegaly and anemia after IFN-γ infusion. Uninfected WT mice were infused with IFN-γ (or phosphate-buffered saline) via osmotic pumps (as described previously14) and spleen weights and blood hemoglobin levels were assessed 6 days later. Mice were simultaneously treated with saline, ruxolitinib (90 mg/kg, twice daily), or anti-IFN-γ antibody as indicated. (D) Survival and clinical score after LCMV infection of prf−/− mice treated with ruxolitinib administered continuously in the drinking water (0.5 mg/mL, delivering 45 mg/kg per day). (E) Assessment of IFN-γ signaling blockade in vivo by continuously administered ruxolitinib. Mice received ruxolitinib in the drinking water (0.5 mg/mL) for 12 hours and were assessed as described in B. N = 5-39 mice/group (39 mice total treated with ruxolitinib in panel A) combined from 3 to 12 independent experiments. *P < .05, **P < .01, ***P < .001.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/12/10.1182_blood.2020007930/4/m_bloodbld2020007930r2f1.png?Expires=1767791125&Signature=2hdS7UH-Yr~FYFfy9-XfU2iyLyJUdSVRzwfxjIpK-H9WFOyrqm4vXVLtGOdBVFecFWqFUWnAT3zJ22kj9ZfhJWScvphnCs~NXvf~WJnEMvkbi1oLD-E4xhdRGsBOd321crC6KQNeUoHu8sfW~SLmIZ3o9YkF~WS~DBNX4d0hflu4s64MF7scfwA7LMFvyQ9a088vJSut0Nc9~~GRq4BmtwKu0V8sfujYTs4fkk0ysPOjQRgilXul2YNGf9EevQRTh~UhjeHS2TpEU75HCxd3qNcaYFszHxS3nexPTODv13RboHixm95SbOlVc8Xr3oZROPLujPdl8P98aKnHQoK8rA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)