

TO THE EDITOR:
Inborn errors of immunity that present with concomitant immunodeficiency and autoinflammation are therapeutically challenging, as recurrent infections limit possibilities of immunosuppression.
The ubiquitin-proteasome system is the main intracellular proteolytic machinery, and mutations in proteasome subunits resulting in proteasome deficiency cause a severe autoinflammatory disease characterized by chronic autoinflammation, fever, neutrophilic dermatosis, and lipodystrophy, collectively known as proteasome-associated autoinflammatory syndromes (PRAAS).1-7 POMP is a chaperone for proteasome assembly, and AD mutations in POMP cause a form of PRAAS with prominent immunodeficiency referred to as POMP-related autoinflammation and immune dysregulation (PRAID). In all forms of PRAAS, autoinflammation is associated with a markedly increased type I interferon response that can be modulated with janus kinase (JAK) inhibitors; however, this therapeutic strategy is not curative, commits patients to lifelong immunosuppression, and is limited by recurrent infections that affect PRAID patients.8-10 Because proteasomes function also in nonimmune cells, it is unclear if restoring the immune system via hematopoietic stem cell transplant (HSCT) will be curative for all aspects of disease, and HSCT has not been previously described for patients with PRAAS. We describe the basis for and outcome of HSCT in 2 PRAID patients in which autoinflammation and immunodeficiency were ameliorated.
Clinical manifestations of both patients were described previously and are detailed in the supplemental Material, available on the Blood Web site.11 Briefly, they suffered from progressive neutrophilic dermatosis, autoimmunity, and combined immunodeficiency characterized by severe early onset, recurrent viral and bacterial infections as well as opportunistic infections.11-13 Immunosuppressive strategies used in both patients to control autoinflammation were ineffective and were associated with multiple intercurrent infections. Although ruxolitinib (JAK1/JAK2 inhibitor) decreased the interferon signature in vitro (supplemental Figure 1A-C), it was considered to present excessive risk in the context of severe Pneumocystis jirovecii infection in patient A and disseminated mycobacterial disease in patient B.
Infectious susceptibility was associated with alterations in T-cell distribution and function characterized by CD4+ naive T-cell skewing with low CD8+ T-cell proportions and decreased cytokine secretion in both CD4+ and CD8+ T cells.11 Although this could partially explain increased infectious susceptibility, we hypothesized that, because POMP is a chaperone for the assembly of all proteasomes, clinical manifestations could be further explained by a defect in thymoproteasomes leading to an impairment in both major histocompatibility complex class I–dependent CD8+ T-cell differentiation and positive antigen selection during thymic development, skewing the T-cell receptor-β (TRB) repertoire.14-18 To investigate the impact of POMP deficiency on T-cell repertoire diversity and composition, we performed high-throughput sequencing to analyze TRB diversity and CDR3 characteristics. As expected, PRAID patients showed a trend toward lower repertoire diversity and increased clonality for all analyzed T-cell subsets (statistically significant for only regulatory T cells [Treg]), and restriction in the number of unique TRB V-J gene pairings identified (Figure 1A-B; supplemental Figure 1D-E). Interestingly, patient A, who had a more profound immunodeficient and autoimmune phenotype, showed increased clonality of CD8+ T cells compared with patient B. These T-cell abnormalities likely represent major drivers of disease and helped favor the decision to proceed with HSCT.
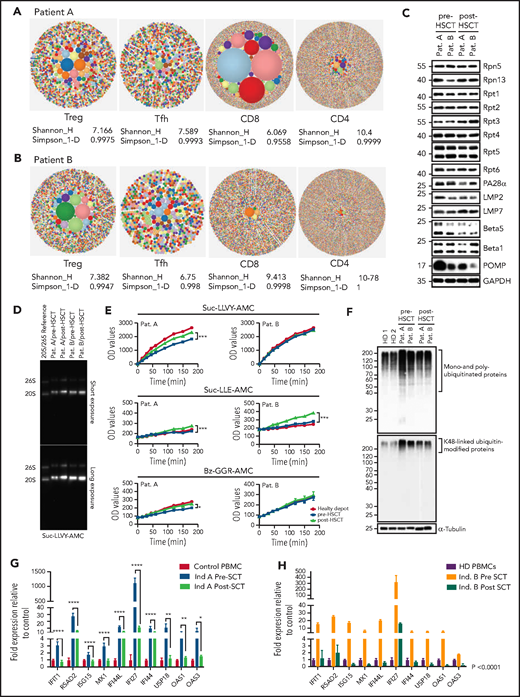
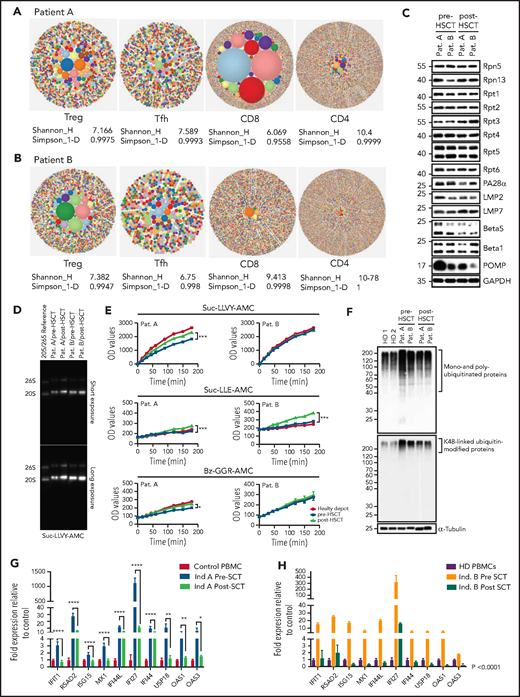
T-cell compartments, proteasome assembly, and function are restored posttransplant, enabling intracellular protein homeostasis and downregulation of the type-1 interferon response. (A-B) TCRb repertoire was determined by high-throughput sequencing of sorted T-cell subsets (T conventional, CD8+ T cells, Treg, T follicular helper cells). Representative hierarchical tree maps show TRB repertoire diversity in patients with truncating POMP mutations. Each dot represents a unique CDR-3, and the size of each dot corresponds to the frequency of that CDR-3 in the total population of sequences obtained. Shannon’s H entropy index measures the diversity of the repertoire, taking into account the clonal size distribution in the overall repertoire. Gini-Simpson index (Simpson_1-D) measures inequality of a given repertoire so that the lower the Simpson_1-D, the more unequal is the distribution of individual clonotypes in the sample results for patients A and B. (C) Whole-cell lysates from pre- and post-SCT peripheral blood mononuclear cell (PBMC) from PRAID patients A and B were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis prior to western blotting using antibodies specific for various proteasome components and/or subunits, including USP14, Rpt1, Rpt2, Rpt3, Rpt4, Rpt5, Rpt6, PA28-α, β1i/LMP2/PSMB9, β5i/LMP7/PSMB10, β5/PSMB5, β1/PSMB6, and POMP, as indicated. Equal protein loading was ensured by probing the membrane with a monoclonal anti-GAPDH antibody. (D) PBMC derived from PRAID patients A and B before and after SCT were subjected to nondenaturing protein extraction to generate protein lysates, which were subsequently run on 3% to 12% native–polyacrylamide gel electrophoresis with proteasome bands being detected by their ability to cleave the Suc-LLVY-AMC fluorogenic peptide. Two exposure times are shown. (E) Protein lysates were tested for their chymotrypsin-, caspase-, and trypsin-like activities by incubating the samples with 0.1 mM of the Suc-LLVY-AMC, Suc-LLE-AMC, and Bz-GGR-AMC fluorogenic substrates, respectively. Fluorescence initiated by AMC release was measured every 15 minutes for the first 2 hours and every 30 minutes for the last 2 hours. Indicated on the y-axis are the raw fluorescence values. (F) Protein lysates derived from patients PBMC pre- and post-SCT were probed for the amounts of K-48–linked ubiquitinated proteins. (G-H) The expression of interferon type-I–inducible genes was evaluated in patient-derived PBMC pre- and posttransplant, and a subset of these was reevaluated posttransplant. The expression of all interferon type-I–regulated gene levels evaluated decreased posttransplant; results are expressed as fold change with respect to control gene expression. Results pretransplant are representative of 3 independent repeats with 3 replicates each. Results posttransplant are only representative of 2 independent repeats with 3 replicates each. Samples were compared using Student t test. Ind., individual; OD, optical density; Pat., patient; SCT, stem cell transplant.
T-cell compartments, proteasome assembly, and function are restored posttransplant, enabling intracellular protein homeostasis and downregulation of the type-1 interferon response. (A-B) TCRb repertoire was determined by high-throughput sequencing of sorted T-cell subsets (T conventional, CD8+ T cells, Treg, T follicular helper cells). Representative hierarchical tree maps show TRB repertoire diversity in patients with truncating POMP mutations. Each dot represents a unique CDR-3, and the size of each dot corresponds to the frequency of that CDR-3 in the total population of sequences obtained. Shannon’s H entropy index measures the diversity of the repertoire, taking into account the clonal size distribution in the overall repertoire. Gini-Simpson index (Simpson_1-D) measures inequality of a given repertoire so that the lower the Simpson_1-D, the more unequal is the distribution of individual clonotypes in the sample results for patients A and B. (C) Whole-cell lysates from pre- and post-SCT peripheral blood mononuclear cell (PBMC) from PRAID patients A and B were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis prior to western blotting using antibodies specific for various proteasome components and/or subunits, including USP14, Rpt1, Rpt2, Rpt3, Rpt4, Rpt5, Rpt6, PA28-α, β1i/LMP2/PSMB9, β5i/LMP7/PSMB10, β5/PSMB5, β1/PSMB6, and POMP, as indicated. Equal protein loading was ensured by probing the membrane with a monoclonal anti-GAPDH antibody. (D) PBMC derived from PRAID patients A and B before and after SCT were subjected to nondenaturing protein extraction to generate protein lysates, which were subsequently run on 3% to 12% native–polyacrylamide gel electrophoresis with proteasome bands being detected by their ability to cleave the Suc-LLVY-AMC fluorogenic peptide. Two exposure times are shown. (E) Protein lysates were tested for their chymotrypsin-, caspase-, and trypsin-like activities by incubating the samples with 0.1 mM of the Suc-LLVY-AMC, Suc-LLE-AMC, and Bz-GGR-AMC fluorogenic substrates, respectively. Fluorescence initiated by AMC release was measured every 15 minutes for the first 2 hours and every 30 minutes for the last 2 hours. Indicated on the y-axis are the raw fluorescence values. (F) Protein lysates derived from patients PBMC pre- and post-SCT were probed for the amounts of K-48–linked ubiquitinated proteins. (G-H) The expression of interferon type-I–inducible genes was evaluated in patient-derived PBMC pre- and posttransplant, and a subset of these was reevaluated posttransplant. The expression of all interferon type-I–regulated gene levels evaluated decreased posttransplant; results are expressed as fold change with respect to control gene expression. Results pretransplant are representative of 3 independent repeats with 3 replicates each. Results posttransplant are only representative of 2 independent repeats with 3 replicates each. Samples were compared using Student t test. Ind., individual; OD, optical density; Pat., patient; SCT, stem cell transplant.
Inflammatory lung involvement in patient A limited HSCT, and the CTLA-4 agonist, abatacept, was used to downregulate activated T cells while reducing risk of immunosuppression. After a 3-month course, pulmonary infiltrates were improved, enabling the initiation of HSCT (supplemental Figure 1F). HSCT was performed at 40 and 43 months of age, in patients A and B, respectively. Patient B was maintained on 4-drug antimycobacterial therapy throughout the process. Both patients were cytomegalovirus seropositive and transplanted with bone marrow from 10 of 10 matched cytomegalovirus-positive unrelated donors. By day 16, both patients had neutrophil and platelet engraftment, and DNA analysis demonstrated 100% donor engraftment of both cell types on days 42 and 100 and 1 year after transplant. Both patients developed mild acute and chronic graft-versus-host disease easily controlled with steroids. Interestingly, despite the use of cyclosporine and steroids, both suffered severe engraftment syndrome starting on day 8 after stem cell infusion (supplemental Table 2). It is possible that elevated interferons in PRAID could underlie increased cytokine release during engraftment,19 and expectant management of this obstacle should be considered.
POMP is critical for β-subunit incorporation into proteasomes, and this process was impaired in POMP-deficient patient cells accounting for accumulation of ubiquitinated proteins and subsequent endoplasmic reticulum stress and exacerbated interferon response.11 Post-HSCT evaluation of proteasome assembly identified increased expression of β5i/LMP7/PSMB8 immunoproteasome subunits with a parallel drop of the β5/PSMB5 standard subunit, suggesting a restoration of proteasome assembly. This correlated with a substantial reduction of POMP expression, likely because of restored β5i/LMP7/PSMB8 promoting physiologic POMP degradation.20 As expected, Rpt1-6 POMP-independent 19S regulatory subunits remained unchanged (Figure 1C). In line with these results, proteasome function analysis showed a significant improvement in chymotrypsin-, caspase-, and trypsin-like activity in patient A, whereas patient B, who had a milder impairment of baseline peptidase activity, only showed statistically significant upregulation for caspase-like activity (Figure 1D-E). We speculate that the specific interpatient differences in POMP protein sequence could differentially impair proteasome function and account for pre-HSCT differences in catalytic activity and disease severity. Importantly, improvement in proteasome function led to significant reduction of intracellular pools of ubiquitin-modified proteins and downregulation of the inherently dysregulated interferon response, indicating that restoration of protein homeostasis in immune cells was sufficient to mitigate systemic interferonopathy (Figure 1F-H). Moreover, these biochemical changes in patient cells correlated with clinical improvement in both patients. Neither had signs of autoinflammation, autoimmunity, or need for medications for these conditions after HSCT. Although patient A retained mild upregulation of some type 1 interferon genes (likely driven by aberrant proteasomes in nonhematopoietic cells), he did not demonstrate active disease.
Immune evaluation 10 to 12 months after transplant showed normalization of CD8+, CD4+, and memory T-cell proportions and restoration of T-cell cytokine secretion. Decreased Treg proportions in patient A also normalized posttransplant (supplemental Figure 2A). Remarkably, T-cell repertoire also improved post-HSCT; patient A showed improvement in diversity and clonality for all T-cell subsets, most significantly in CD8+ T cells, and patient B showed improved Treg clonality while diversity of other subsets remained unchanged at the time of evaluation (Figure 2E). Both patients showed an improvement in Treg V-J gene pair usage (supplemental Figure 2B-C). These results demonstrated HSCT was sufficient to rescue T-cell compartments and differentiation in PRAID patients despite presumed persistent POMP aberrancies in thymoproteasomes of cortical thymic epithelial cells, potentially underscoring the role of donor dendritic cells that repopulate the thymus after HSCT in promoting both Treg and CD8+ T-cell differentiation.
![T-cell compartments and T-cell cytokine production are restored after hematopoietic stem cell transplant. (A) T-cell subsets evaluated before transplant show a decreased proportion of CD8+ T cells and increased proportion of CD4+ T cells compared with control that increase and decrease, respectively, posttransplant to proportions that are comparable to control T cells. The figure is representative of 3 independent repeats pre- and posttransplant. (B) T-cell memory subsets before transplant show an increased proportion of naive T cells (CCR7hi/CD45ROLo) and a reduced proportion of effector and effector memory T cells (CCR7lo/CD45ROhi and CCR7lo/CD45ROlo). (C) Cytokine secretion (interferon-γ [IFN-γ], interleukin-2 [IL-2], and tumor necrosis factor-α [TNF-α]) in pre- and posttransplant CD8+ T cells. Graphs show percent of positive cells after staphylococcal enterotoxin B stimulation, normalized to control for each cytokine. Cells were gated on live cells, CD3+, CD4+, and CD8+ T cells, and percentage of cytokine secreting cells was determined against side scatter for each subset. Cytokine secretion was measured on 3 separate time points before transplant and at least twice posttransplant. (D) Cytokine secretion in CD4+ T cells pre- and posttransplant. Graphs show percent of positive cells after staphylococcal enterotoxin B stimulation normalized to control for each cytokine. Cells were gated on live cells, CD3+, CD4+, and CD8+ T cells, and percentage of cytokine-secreting cells was determined against side scatter for each subset. Cytokine secretion was measured on 3 separate timepoints before transplant and at least twice posttransplant. (E) TCRb repertoire was determined by high-throughput sequencing of sorted T-cell subsets (T conventional, CD8+ T cells, Treg, T follicular helper cells). Representative hierarchical tree maps show TRB repertoire diversity in patient PBMC samples after transplant. Each dot represents a unique CDR-3, and the size of each dot corresponds to the frequency of that CDR-3 in the total population of sequences obtained. Shannon’s H entropy index measures the diversity of the repertoire, taking into account the clonal size distribution in the overall repertoire, and is indicated below each tree map.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/19/10.1182_blood.2021011005/4/m_bloodbld2021011005f2.png?Expires=1769172387&Signature=sTv9K7hZwuNq5fmcfcyoClkKWOY6yqeF4fex8HbFfguuvyVM8roB4axtR8GoHoRJ10y6givSDs~hnGz3Fw~bXaL1c5SDTTXW9g8CJ1oJNLGBnBqUjR1fctjTtEzdkA9tXVpU3TgU2W35xY08hC1jzsW0o1~~djhfjK8A6J9Sozirn70rSBUELR8Tkg2Cw2n9pw-2D6MdO-NAWl6aM9IvFFMCOHFS4WflDZI3vvRq0aA5bZgRIve-5Zs0D-8jiUS7I30JA~F62c-L35cz4xd4zWV0YyXdsXkbT1zzIQh0Bct~wlCa7-OBOkIfQTz7tEMl0XA4X17fpl40-f03-7DV0g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
T-cell compartments and T-cell cytokine production are restored after hematopoietic stem cell transplant. (A) T-cell subsets evaluated before transplant show a decreased proportion of CD8+ T cells and increased proportion of CD4+ T cells compared with control that increase and decrease, respectively, posttransplant to proportions that are comparable to control T cells. The figure is representative of 3 independent repeats pre- and posttransplant. (B) T-cell memory subsets before transplant show an increased proportion of naive T cells (CCR7hi/CD45ROLo) and a reduced proportion of effector and effector memory T cells (CCR7lo/CD45ROhi and CCR7lo/CD45ROlo). (C) Cytokine secretion (interferon-γ [IFN-γ], interleukin-2 [IL-2], and tumor necrosis factor-α [TNF-α]) in pre- and posttransplant CD8+ T cells. Graphs show percent of positive cells after staphylococcal enterotoxin B stimulation, normalized to control for each cytokine. Cells were gated on live cells, CD3+, CD4+, and CD8+ T cells, and percentage of cytokine secreting cells was determined against side scatter for each subset. Cytokine secretion was measured on 3 separate time points before transplant and at least twice posttransplant. (D) Cytokine secretion in CD4+ T cells pre- and posttransplant. Graphs show percent of positive cells after staphylococcal enterotoxin B stimulation normalized to control for each cytokine. Cells were gated on live cells, CD3+, CD4+, and CD8+ T cells, and percentage of cytokine-secreting cells was determined against side scatter for each subset. Cytokine secretion was measured on 3 separate timepoints before transplant and at least twice posttransplant. (E) TCRb repertoire was determined by high-throughput sequencing of sorted T-cell subsets (T conventional, CD8+ T cells, Treg, T follicular helper cells). Representative hierarchical tree maps show TRB repertoire diversity in patient PBMC samples after transplant. Each dot represents a unique CDR-3, and the size of each dot corresponds to the frequency of that CDR-3 in the total population of sequences obtained. Shannon’s H entropy index measures the diversity of the repertoire, taking into account the clonal size distribution in the overall repertoire, and is indicated below each tree map.
T-cell compartments and T-cell cytokine production are restored after hematopoietic stem cell transplant. (A) T-cell subsets evaluated before transplant show a decreased proportion of CD8+ T cells and increased proportion of CD4+ T cells compared with control that increase and decrease, respectively, posttransplant to proportions that are comparable to control T cells. The figure is representative of 3 independent repeats pre- and posttransplant. (B) T-cell memory subsets before transplant show an increased proportion of naive T cells (CCR7hi/CD45ROLo) and a reduced proportion of effector and effector memory T cells (CCR7lo/CD45ROhi and CCR7lo/CD45ROlo). (C) Cytokine secretion (interferon-γ [IFN-γ], interleukin-2 [IL-2], and tumor necrosis factor-α [TNF-α]) in pre- and posttransplant CD8+ T cells. Graphs show percent of positive cells after staphylococcal enterotoxin B stimulation, normalized to control for each cytokine. Cells were gated on live cells, CD3+, CD4+, and CD8+ T cells, and percentage of cytokine secreting cells was determined against side scatter for each subset. Cytokine secretion was measured on 3 separate time points before transplant and at least twice posttransplant. (D) Cytokine secretion in CD4+ T cells pre- and posttransplant. Graphs show percent of positive cells after staphylococcal enterotoxin B stimulation normalized to control for each cytokine. Cells were gated on live cells, CD3+, CD4+, and CD8+ T cells, and percentage of cytokine-secreting cells was determined against side scatter for each subset. Cytokine secretion was measured on 3 separate timepoints before transplant and at least twice posttransplant. (E) TCRb repertoire was determined by high-throughput sequencing of sorted T-cell subsets (T conventional, CD8+ T cells, Treg, T follicular helper cells). Representative hierarchical tree maps show TRB repertoire diversity in patient PBMC samples after transplant. Each dot represents a unique CDR-3, and the size of each dot corresponds to the frequency of that CDR-3 in the total population of sequences obtained. Shannon’s H entropy index measures the diversity of the repertoire, taking into account the clonal size distribution in the overall repertoire, and is indicated below each tree map.
Most importantly, both patients encountered viral infections after transplant without requiring aggressive interventions, confirming appropriate reconstitution of their immunity. Both patients are currently alive and well at 56- and 32-months posttransplant, respectively, with no signs of autoinflammation, autoimmunity, lipodystrophy, or recurrent infections (supplemental Table 2).
Overall, we demonstrate HSCT is an effective therapeutic strategy for PRAID patients. Our results highlight that POMP rescue in hematopoietic cells is sufficient to significantly downregulate systemic interferonopathy, autoinflammation, and immunodeficiency, despite the continued extraimmune cell expression of aberrant POMP protein, suggesting interferonopathy in PRAID is driven by proteasome deficiency in immune cells. To our knowledge, these are the first patients with a form of PRAAS that have been successfully transplanted, representing a promising therapeutic direction for patients with POMP deficiency and other forms PRAAS.21,22
Acknowledgments
The authors acknowledge the following clinical consult physicians who participated in patient care: Ryan Himes, gastroenterology; Timothy Vece, pulmonology; Carl Allen, Malcom Brenner and Helen Heslop hematology oncology; Regan Hunt, dermatology; Alisa Acosta, nephrology; and Stacey Shubert for patient care. The authors are especially thankful to both patients described in this article and their families for facilitating this work. The authors are grateful to Malcom Brenner for critically reviewing the manuscript and providing insightful advice.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
This work was supported, in part, by FONDECYT grant 11181222 (M.C.P.), National Institutes of Health (NIH), National Institute of Allergy and Infectious Diseases grant R01AI120989 (J.S.O.), Fritz Thyssen Foundation grant AZ:10.16.2.022 MN (E.K.), and German Research Foundation grant SFBTRR 167 A04 (E.K.). L.D.N. is supported by the NIH, Division of Intramural Research, National Institute of Allergy and Infectious Diseases.
Authorship
Contribution: C.M. performed and supervised HSCT; C.M., M.C.P., and F.E. wrote the manuscript; M.C.P., F.E., M.B., R.C., and O.M.D. performed and analyzed experiments; M.C.P. was responsible for study design; L.D.N. and E.K. supervised data interpretation and critically revised the manuscript; L.R.F. critically reviewed the manuscript. L.R.F., S.K.N., M.D.G., and R.K. were involved in patient care; J.S.O. supervised data interpretation, was involved in study design, and critically revised the manuscript; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: M. Cecilia Poli, Instituto de Ciencias e Innovación en Medicina, Facultad de Medicina Clínica Alemana-Universidad del Desarrollo, Av Las Condes 12.461 Of. 205, Santiago, Chile 7590943; e-mail: cpoli@udd.cl or maria.poli@bcm.edu.
There is a Blood Commentary on this article in this issue.
The online version of this article contains a data supplement.

![T-cell compartments and T-cell cytokine production are restored after hematopoietic stem cell transplant. (A) T-cell subsets evaluated before transplant show a decreased proportion of CD8+ T cells and increased proportion of CD4+ T cells compared with control that increase and decrease, respectively, posttransplant to proportions that are comparable to control T cells. The figure is representative of 3 independent repeats pre- and posttransplant. (B) T-cell memory subsets before transplant show an increased proportion of naive T cells (CCR7hi/CD45ROLo) and a reduced proportion of effector and effector memory T cells (CCR7lo/CD45ROhi and CCR7lo/CD45ROlo). (C) Cytokine secretion (interferon-γ [IFN-γ], interleukin-2 [IL-2], and tumor necrosis factor-α [TNF-α]) in pre- and posttransplant CD8+ T cells. Graphs show percent of positive cells after staphylococcal enterotoxin B stimulation, normalized to control for each cytokine. Cells were gated on live cells, CD3+, CD4+, and CD8+ T cells, and percentage of cytokine secreting cells was determined against side scatter for each subset. Cytokine secretion was measured on 3 separate time points before transplant and at least twice posttransplant. (D) Cytokine secretion in CD4+ T cells pre- and posttransplant. Graphs show percent of positive cells after staphylococcal enterotoxin B stimulation normalized to control for each cytokine. Cells were gated on live cells, CD3+, CD4+, and CD8+ T cells, and percentage of cytokine-secreting cells was determined against side scatter for each subset. Cytokine secretion was measured on 3 separate timepoints before transplant and at least twice posttransplant. (E) TCRb repertoire was determined by high-throughput sequencing of sorted T-cell subsets (T conventional, CD8+ T cells, Treg, T follicular helper cells). Representative hierarchical tree maps show TRB repertoire diversity in patient PBMC samples after transplant. Each dot represents a unique CDR-3, and the size of each dot corresponds to the frequency of that CDR-3 in the total population of sequences obtained. Shannon’s H entropy index measures the diversity of the repertoire, taking into account the clonal size distribution in the overall repertoire, and is indicated below each tree map.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/19/10.1182_blood.2021011005/4/m_bloodbld2021011005f2.png?Expires=1769172388&Signature=n1YL0PeiHDkj-w7Tl6LApRlUcYiGMcMwc9c8cW5GaYWsfeIbghtLPIptVx3kw2VDEPXO4DKJZfFV33w38FAX8cK0gRfmRMrBuWn9fn~ACd-gZl5FwbwRFamU~MTsXH8-iB37tsZpzEN8Mj0etkPRGoZLU6eB-Km7Il229kIKc0~TibsHpJ-xHA8ngorJ5iqzmszj0f60-bm7D79sM3DNHVe30mNROlXmVt0-RnOnDcjG-D6AZ65i1nBgwS6xBgxt-OorlFEZxat2mF7Whoc3BnvfaCoRzweYBO8P~jELW8ehj~buShiYH6dqq2NuKe4pG7H1j33UiH1ZSz5tl2vKLg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
