

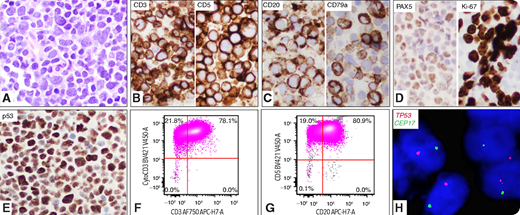
A 59-year-old woman presented with lymphadenopathies. A lymph node biopsy revealed intermediate- to large-sized blastoid cells with round to irregular nuclei, fine chromatin, and small inconspicuous nucleoli (panel A; original magnification: ×40 objective; ×400 total magnification). By immunohistochemistry, the neoplastic cells were positive for CD3, CD5, CD20, CD79a, PAX5, Ki67, and p53 (panels B-E; original magnification: ×40 objective; ×400 total magnification). Flow cytometric analysis showed aberrant T cells: positive for CD2, CD3 (panel F; surface and cytoplasmic), CD4, CD5, CD20 (panel G), TCRα/β, and negative for CD34, TDT, CD1a, CD7, CD8, CD10, CD30, TCRγ/δ, CD19, and CD22. Molecular studies detected monoclonal TCR β and γ gene rearrangements but no IGH rearrangement. Fluorescence in situ hybridization using dual-color TP53/CEP17 probes detected monosomy 17 in 88% of cells (panel H; original magnification: ×60 objective; ×600 total magnification). A 162-gene lymphoma mutation assay by next-generation sequencing revealed TP53 mutation (variant allele frequency = 80%). She was diagnosed with peripheral T-cell lymphoma (PTCL), not otherwise specified, and was treated with chemotherapy with poor response. She died of disease in 5 months.
PTCL with blastoid morphology and multiple B-cell markers is highly unusual. The blastoid morphology raises concern for a lymphoblastic lymphoma, and extensive workup is critical in reaching the correct diagnosis. Biallelic TP53 loss of function mostly likely contributed to the patient’s lymphomagenesis and dismal outcome.
A 59-year-old woman presented with lymphadenopathies. A lymph node biopsy revealed intermediate- to large-sized blastoid cells with round to irregular nuclei, fine chromatin, and small inconspicuous nucleoli (panel A; original magnification: ×40 objective; ×400 total magnification). By immunohistochemistry, the neoplastic cells were positive for CD3, CD5, CD20, CD79a, PAX5, Ki67, and p53 (panels B-E; original magnification: ×40 objective; ×400 total magnification). Flow cytometric analysis showed aberrant T cells: positive for CD2, CD3 (panel F; surface and cytoplasmic), CD4, CD5, CD20 (panel G), TCRα/β, and negative for CD34, TDT, CD1a, CD7, CD8, CD10, CD30, TCRγ/δ, CD19, and CD22. Molecular studies detected monoclonal TCR β and γ gene rearrangements but no IGH rearrangement. Fluorescence in situ hybridization using dual-color TP53/CEP17 probes detected monosomy 17 in 88% of cells (panel H; original magnification: ×60 objective; ×600 total magnification). A 162-gene lymphoma mutation assay by next-generation sequencing revealed TP53 mutation (variant allele frequency = 80%). She was diagnosed with peripheral T-cell lymphoma (PTCL), not otherwise specified, and was treated with chemotherapy with poor response. She died of disease in 5 months.
PTCL with blastoid morphology and multiple B-cell markers is highly unusual. The blastoid morphology raises concern for a lymphoblastic lymphoma, and extensive workup is critical in reaching the correct diagnosis. Biallelic TP53 loss of function mostly likely contributed to the patient’s lymphomagenesis and dismal outcome.
For additional images, visit the ASH Image Bank, a reference and teaching tool that is continually updated with new atlas and case study images. For more information, visit http://imagebank.hematology.org.

