Key Points
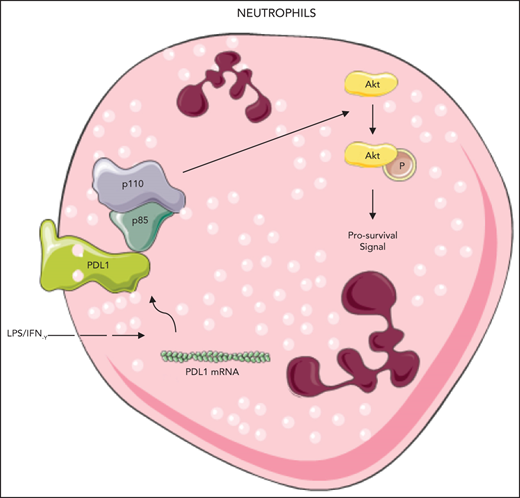
PD-L1 upregulation prolongs septic neutrophil cell survival by activating PI3K/AKT signaling.
Neutrophil-specific PD-L1 knockdown attenuates lung injury and improves mortality in mice challenged with cecal ligation and puncture.

Abstract
PD-L1 is a ligand for PD-1, and its expression has been shown to be upregulated in neutrophils harvested from septic patients. However, the effect of PD-L1 on neutrophil survival and sepsis-induced lung injury remains largely unknown. In this study, PD-L1 expression correlated negatively with rates of apoptosis in human neutrophils harvested from patients with sepsis. Coimmunoprecipitation assays on control neutrophils challenged with interferon-γ and LPS showed that PD-L1 complexes with the p85 subunit of phosphatidyl 3-kinase (PI3K) to activate AKT-dependent survival signaling. Conditional CRE/LoxP deletion of neutrophil PD-L1 in vivo further protected against lung injury and reduced neutrophil lung infiltration in a cecal ligation and puncture (CLP) experimental sepsis animal model. Compared with wild-type animals, PD-L1–deficient animals presented lower levels of plasma tumor necrosis factor-α and interleukin-6 (IL-6) and higher levels of IL-10 after CLP, and reduced 7-day mortality in CLP PD-L1–knockout animals. Taken together, our data suggest that increased PD-L1 expression on human neutrophils delays cellular apoptosis by triggering PI3K–dependent AKT phosphorylation to drive lung injury and increase mortality during clinical and experimental sepsis.
Introduction
Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection and is the leading cause of death in the intensive care unit.1,2 Treatment options for sepsis remain limited by an incomplete understanding of the molecular mechanisms driving the host response to infection. Multiple studies have indicated that patients with sepsis experience disordered immune function.3,4 Our previous studies have demonstrated that sustained neutrophil activation through delayed apoptosis contributes to nonspecific tissue injury in patients with sepsis.5,6
PD-L1 is a ligand for PD-1 and negatively regulates T-cell responses by inducing lymphocyte apoptosis. The expression of PD-L1, a coinhibitory molecule, is increased in neutrophils during sepsis.7,8 We have shown that PD-L1 is upregulated in neutrophils and correlates with sepsis-induced immunosuppression.8 Other work has shown that neutrophil apoptosis is regulated through the phosphatidyl 3–kinase (PI3K)/AKT signaling pathway,9,10 and it has been reported that PD-L1 maintains the stemness of cancer stem cells through AKT signaling.11 We therefore sought to investigate whether PD-L1 delays ex vivo neutrophil apoptosis by inducing AKT phosphorylation in neutrophils extrapolated from consenting healthy volunteers. We further identified the in vivo role of neutrophil PD-L1 in sepsis by using a cecal ligation and puncture experimental animal model.
Study design
Patients
Patients who fulfilled the clinical criteria for sepsis12,13 were recruited from the Medical Surgical Intensive Care Unit of St Michael’s Hospital and the Central Intensive Care Unit of Changhai Hospital. Consenting healthy donors served as controls. The study protocols were approved by the Human Research Ethics Board of St Michael’s Hospital (Toronto, ON, Canada) and the Committee on Ethics of Biomedicine Research, Naval Medical University (Shanghai, China).
Mice
Neutrophil-specific, PD-L1 conditioned, knockout mice were generated by breeding PD-L1flox/flox, engineered using CRISPR/Cas9 (Bioray Laboratories Inc, Shanghai, China), with elane (Ela)cre/cre mice14,15 purchased from the EMMA mouse repository (Infrafrontier, Munich, Germany). The specificity of PD-L1 knockout in mice is shown in supplemental Figure 1, available on the Blood Web site. All animal studies were approved by the Committee on Ethics of Biomedicine Research in Naval Medical University, Shanghai, China.
Cecal ligation and puncture experiments
Sepsis was induced in C57/Bl6 animals by surgically performing cecal ligation and puncture (CLP), as described previously.8 Hematoxylin and eosin staining was conducted to quantify lung injury.16 Neutrophil accumulation in the lung was determined by quantifying fluorescent MPO+ cells. Plasma levels of tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) and -10 were detected by enzyme-linked immunosorbent assay (R&D, Minneapolis, MN). The survival rate was observed for 7 days after CLP surgery.
Neutrophil purification, transfection, and apoptosis assay
Human neutrophils were purified by density gradient centrifugation with 3% dextran and Ficoll-Hypaque (GE Healthcare, Little Chalfont, United Kingdom), as previously described.5,6 The purity of neutrophils is shown in supplemental Figure 2.
More details can be found in the supplementary Methods.
Results and discussion
Interferon-γ (IFN-γ) induces PD-L1 expression,17 and LPS delays neutrophil apoptosis.5,6 Challenging healthy human neutrophils with IFN-γ and/or LPS induced PD-L1 expression and significantly delayed neutrophil apoptosis at 21 hours after stimulation (Figure 1A). PD-L1 expression was increased in neutrophils isolated from patients with sepsis and healthy volunteers and correlated negatively with apoptosis rates in septic neutrophils 21 hours after isolation (Figure 1B). To determine whether IFN-γ and LPS-induced PD-L1 expression contributes to delayed neutrophil apoptosis, we genetically silenced PD-L1 expression with small interfering RNA (siRNA). After confirming transfection by using flow cytometry, PD-L1–deficient human control neutrophils stimulated with IFN-γ and LPS exhibited significantly higher rates of apoptosis (Figure 1C). Similar findings were replicated in human septic neutrophils transfected with PD-L1 siRNA (Figure 1D; supplemental Figures 3 and 4). To further validate the flow cytometry results, we conducted western blot experiments to measure the protein levels of cleaved caspase-3. Human healthy control neutrophils stimulated with IFN-γ and LPS demonstrated lower cleaved caspase-3 expression than the untreated neutrophils showed (supplemental Figure 5). Similar trends were observed in neutrophils isolated from the bone marrow (BM) of mice, where PD-L1 expression correlated negatively with rates of apoptosis (supplemental Figure 6A-B). In aggregate, these data suggest that PD-L1 contributes to delayed neutrophil apoptosis in patients with sepsis.
![Increased PD-L1 expression in activated neutrophils inhibits cellular apoptosis by maintaining AKT phosphorylation. (A) PD-L1 expression was increased and rates of apoptosis were significantly decreased in human healthy control neutrophils stimulated with IFN-γ (10 ng/mL) and/or LPS (1 μg/mL) for 21 hours (n = 6; *P < .05, 1-way analysis of variance [ANOVA]). (B) Twenty-one hours after isolation, PD-L1 expression was increased in neutrophils from patients with sepsis, when compared with resting control neutrophils, and correlated negatively with rates of apoptosis (n = 14; P < .05, by linear regression analysis). (C) Genetic silencing of PD-L1 using siRNA in human control neutrophils stimulated with IFN-γ (10 ng/mL) and/or LPS (1 μg/mL) for 21 hours significantly increased apoptosis (n = 5). *P < .05, 1-way ANOVA. (D) Twenty-one hours after isolation, human septic neutrophils transfected with PD-L1 siRNA demonstrated significantly higher rates of apoptosis than those transfected with scrambled control siRNA (n = 5). *P < .05, 1-way ANOVA. (E) Representative immunoblot shows that AKT phosphorylation (S473) is increased in human healthy control neutrophils challenged with IFN-γ (10 ng/mL) and LPS (1 μg/mL; n = 3) 21 hours after isolation. (F) Representative immunoblots measuring AKT phosphorylation in neutrophil lysates from healthy donors vs that in patients with sepsis (n = 3). The representative blots for PD-L1 and β-actin correspond to the data points shown in panel B. (G) Genetic silencing of PD-L1 in human septic neutrophils with siRNA decreased AKT phosphorylation 21 hours after isolation (n = 3). (H) HEK293 cells were transfected with empty GFP plasmid or GFP-tagged PD-L1. Immunoprecipitated GFP alone and GFP-tagged PD-L1 lysates demonstrate that PD-L1 complexed with p85 (n = 3).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/9/10.1182_blood.2020009417/5/m_bloodbld2020009417f1.png?Expires=1768316450&Signature=JlWy2HLeNtVoCwKLmNx5j7MunwlKjrymIbp6SOZzCaK1aAt8oe81WW4-AKmbJyuQPuRMaxq-phazOsq8szWpa-N1DDmBaSXiXPUURuEzCScIHDvrpFuYEx5PpMDFoz04LW4wgag-4LtCECutLiI3XQNoUNk1LZM2ZLX9fGeNtJBvZ-ikxnpf2taiaVhU~O1xXA9i6sUZF4iJOIQq7d5qSuzx9SUP3sA-nKNFmc6w1XZydJzM2XAmW4KdzVZ-ClOZ1Lel5I4vzqOVICeQu4bZnPwpZaBMXCSmGg6AHE7H~mW5d0WtCJWN4HvorEHwLWJjX7zZ-8YgbqT31oh6rWojXA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Increased PD-L1 expression in activated neutrophils inhibits cellular apoptosis by maintaining AKT phosphorylation. (A) PD-L1 expression was increased and rates of apoptosis were significantly decreased in human healthy control neutrophils stimulated with IFN-γ (10 ng/mL) and/or LPS (1 μg/mL) for 21 hours (n = 6; *P < .05, 1-way analysis of variance [ANOVA]). (B) Twenty-one hours after isolation, PD-L1 expression was increased in neutrophils from patients with sepsis, when compared with resting control neutrophils, and correlated negatively with rates of apoptosis (n = 14; P < .05, by linear regression analysis). (C) Genetic silencing of PD-L1 using siRNA in human control neutrophils stimulated with IFN-γ (10 ng/mL) and/or LPS (1 μg/mL) for 21 hours significantly increased apoptosis (n = 5). *P < .05, 1-way ANOVA. (D) Twenty-one hours after isolation, human septic neutrophils transfected with PD-L1 siRNA demonstrated significantly higher rates of apoptosis than those transfected with scrambled control siRNA (n = 5). *P < .05, 1-way ANOVA. (E) Representative immunoblot shows that AKT phosphorylation (S473) is increased in human healthy control neutrophils challenged with IFN-γ (10 ng/mL) and LPS (1 μg/mL; n = 3) 21 hours after isolation. (F) Representative immunoblots measuring AKT phosphorylation in neutrophil lysates from healthy donors vs that in patients with sepsis (n = 3). The representative blots for PD-L1 and β-actin correspond to the data points shown in panel B. (G) Genetic silencing of PD-L1 in human septic neutrophils with siRNA decreased AKT phosphorylation 21 hours after isolation (n = 3). (H) HEK293 cells were transfected with empty GFP plasmid or GFP-tagged PD-L1. Immunoprecipitated GFP alone and GFP-tagged PD-L1 lysates demonstrate that PD-L1 complexed with p85 (n = 3).
Increased PD-L1 expression in activated neutrophils inhibits cellular apoptosis by maintaining AKT phosphorylation. (A) PD-L1 expression was increased and rates of apoptosis were significantly decreased in human healthy control neutrophils stimulated with IFN-γ (10 ng/mL) and/or LPS (1 μg/mL) for 21 hours (n = 6; *P < .05, 1-way analysis of variance [ANOVA]). (B) Twenty-one hours after isolation, PD-L1 expression was increased in neutrophils from patients with sepsis, when compared with resting control neutrophils, and correlated negatively with rates of apoptosis (n = 14; P < .05, by linear regression analysis). (C) Genetic silencing of PD-L1 using siRNA in human control neutrophils stimulated with IFN-γ (10 ng/mL) and/or LPS (1 μg/mL) for 21 hours significantly increased apoptosis (n = 5). *P < .05, 1-way ANOVA. (D) Twenty-one hours after isolation, human septic neutrophils transfected with PD-L1 siRNA demonstrated significantly higher rates of apoptosis than those transfected with scrambled control siRNA (n = 5). *P < .05, 1-way ANOVA. (E) Representative immunoblot shows that AKT phosphorylation (S473) is increased in human healthy control neutrophils challenged with IFN-γ (10 ng/mL) and LPS (1 μg/mL; n = 3) 21 hours after isolation. (F) Representative immunoblots measuring AKT phosphorylation in neutrophil lysates from healthy donors vs that in patients with sepsis (n = 3). The representative blots for PD-L1 and β-actin correspond to the data points shown in panel B. (G) Genetic silencing of PD-L1 in human septic neutrophils with siRNA decreased AKT phosphorylation 21 hours after isolation (n = 3). (H) HEK293 cells were transfected with empty GFP plasmid or GFP-tagged PD-L1. Immunoprecipitated GFP alone and GFP-tagged PD-L1 lysates demonstrate that PD-L1 complexed with p85 (n = 3).
PI3K is indispensable for the antiapoptotic activity of LPS and for the transcription of proteins that inhibit apoptosis.9,18 We therefore sought to investigate whether increased PD-L1 expression alters activation of PI3K by assessing downstream AKT phosphorylation. AKT phosphorylation was enhanced in resting human neutrophils stimulated with IFN-γ and LPS, as well as neutrophils from patients with sepsis (Figure 1E-F). In PD-L1–knockdown human neutrophils, AKT phosphorylation was reduced, suggesting that PD-L1 maintains PI3K activation (Figure 1G). In murine BM neutrophils challenged with IFN-γ and LPS, PD-L1 expression, and AKT phosphorylation also increased (supplemental Figure 7). Pretreating control human neutrophils with a PI3K inhibitor, LY294002 (10 μM), before IFN-γ and LPS challenge, blocked prosurvival AKT signaling and increased caspase-3 cleavage (supplemental Figure 8). These data suggest that PI3K activation is necessary to prolong neutrophil survival and that PD-L1 may be part of a larger, prosurvival signaling complex controlled by PI3K. This hypothesis is supported by reduced AKT phosphorylation in neutrophil-specific PD-L1–deficient animals (supplemental Figure 9A). Rates of apoptosis were also higher in PD-L1–deficient murine neutrophils challenged with IFN-γ and LPS compared with wild-type controls (supplemental Figure 9B). AKT phosphorylation is dependent on the activation of PI3K through the liberation of the p110 subunit from p85,19,20 and we therefore investigated the interaction between p85 and PD-L1 by immunoprecipitation. Empty GFP plasmid failed to bind to endogenous p85, whereas GFP-tagged PD-L1 bound to p85 in HEK293 cells (Figure 1H). Taken together, these data suggest that PD-L1 interacts with the p85 subunit of PI3K to enable downstream AKT phosphorylation and delay neutrophil apoptosis.
To investigate the role of neutrophil PD-L1 in the pathogenesis of sepsis, we engineered control and neutrophil-specific PD-L1 knockout mice. Plasma levels of TNF-α and IL-6 were significantly lower, and IL-10 level was significantly higher in PD-L1flox/flox CLP mice when compared with wild-type animals. However, the IFN-γ level was similar in both control and PD-L1 knockout CLP mice (Figure 2A-C; supplemental Figure 10). Lung histology revealed a reduction in inflammatory cells and edema fluid within the alveoli and slight interstitial exudation in PD-L1flox/flox CLP mice, which is reflected in improved lung injury scores (Figure 2D). Consistent with the histologic data, there were fewer MPO+ cells in the lungs of PD-L1flox/flox CLP mice when compared with those of PD-L1WT/WT CLP mice (Figure 2E). To investigate whether PD-L1 alters the expression of adhesive and chemotactic molecules, we measured ICAM-1, CXCR2, and CCR2 by flow cytometry. Our data showed no difference in ICAM-1, CXCR2, and CCR2 expression in PD-L1flox/flox and PD-L1WT/WT CLP mice (supplemental Figure 11). These findings suggest that lower neutrophil lung infiltration in PD-L1flox/flox CLP mice is not influenced by adhesion molecule expression and chemotaxis. Finally, mortality rates in PD-L1flox/flox CLP mice at 7 days was significantly lower when compared with those in PD-L1WT/WT CLP mice (Figure 2F).
![Genetic deletion of neutrophil-specific PD-L1 in vivo decreases proinflammatory plasma cytokine expression, thus limiting lung injury and neutrophil infiltration in the lung and decreasing mortality. (A-C) Plasma concentration of TNF-α (A), IL-6 (B), and IL-10 (C) were determined by enzyme-linked immunosorbent assay. Plasma concentrations of TNF-α and IL-6 were significantly reduced in PD-L1flox/flox CLP mice when compared with levels in PD-L1WT/WT CLP mice, but were still higher than in sham-treated PD-L1WT/WT mice or sham-treated PD-L1flox/flox mice. IL-10 concentration was significantly elevated in PD-L1flox/flox CLP mice when compared with PD-L1WT/WT CLP mice (n = 6). *P < .05, 1-way analysis of variance [ANOVA]. (D) Lung injury score and the representative histopathological images of the lungs. PD-L1flox/flox CLP mice demonstrate significantly lower lung injury score when compared with PD-L1WT/WT sham-treated mice, PD-L1flox/flox sham-treated mice, and PD-L1WT/WT CLP mice (scale bar, 100 μm) (n = 5). *P < .05, Kruskal-Wallis test with Dunn’s multiple comparisons test. (E) Representative immunofluorescence images of MPO (green) staining with blue 4′,6-diamidino-2-phenylindole (DAPI) nuclear stain in lung tissue. The percentage of MPO+ cells was significantly lower in PD-L1flox/flox CLP mice when compared with that in PD-L1WT/WT CLP mice, but was slightly higher than that in PD-L1WT/WT mice in the sham-operation group and PD-L1flox/flox mice (scale bar, 50 μm) (n = 5). *P < .05, 1-way ANOVA. (F) After CLP, PD-L1flox/flox CLP mice demonstrated higher 7-day survival rates than PD-L1WT/WT CLP mice (n = 8 for PD-L1WT/WT and PD-L1flox/flox mice in the sham-operation group; n = 18 for PD-L1WT/WT and PD-L1flox/flox CLP mice). *P < .05, by log-rank test.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/9/10.1182_blood.2020009417/5/m_bloodbld2020009417f2.png?Expires=1768316450&Signature=2AIQUuGUqBInOyIDzDvNNAvLdQC-CCV4H-HzvAbaJKxdB3egviQztfDz65iGBakNU7ZIirFdoDIyzApwKYU0~xtdQMcC2aDbGzqY8Ll9calg1tTfdoZb8iVxAUShq-b6RjXMxM2k8lyMgXWgj6in2eeV7grCtmZ4DtPSPlMUkWC47CRPwZoH3h7hrzXVCO9riqkGxkZZcXNo7L6dID39ACoFJYJYs8g-Q6ECySq-ifmn~BRhGp-iDc1QkkKE7ovn0m3dpalCS7zV3aJaCmOJR-ma0utGrpG9fXmsNvO3ZNQltXb1Bxw01zJFD4h36n-hnUFLTBOzJyKrotwgQimYnQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Genetic deletion of neutrophil-specific PD-L1 in vivo decreases proinflammatory plasma cytokine expression, thus limiting lung injury and neutrophil infiltration in the lung and decreasing mortality. (A-C) Plasma concentration of TNF-α (A), IL-6 (B), and IL-10 (C) were determined by enzyme-linked immunosorbent assay. Plasma concentrations of TNF-α and IL-6 were significantly reduced in PD-L1flox/flox CLP mice when compared with levels in PD-L1WT/WT CLP mice, but were still higher than in sham-treated PD-L1WT/WT mice or sham-treated PD-L1flox/flox mice. IL-10 concentration was significantly elevated in PD-L1flox/flox CLP mice when compared with PD-L1WT/WT CLP mice (n = 6). *P < .05, 1-way analysis of variance [ANOVA]. (D) Lung injury score and the representative histopathological images of the lungs. PD-L1flox/flox CLP mice demonstrate significantly lower lung injury score when compared with PD-L1WT/WT sham-treated mice, PD-L1flox/flox sham-treated mice, and PD-L1WT/WT CLP mice (scale bar, 100 μm) (n = 5). *P < .05, Kruskal-Wallis test with Dunn’s multiple comparisons test. (E) Representative immunofluorescence images of MPO (green) staining with blue 4′,6-diamidino-2-phenylindole (DAPI) nuclear stain in lung tissue. The percentage of MPO+ cells was significantly lower in PD-L1flox/flox CLP mice when compared with that in PD-L1WT/WT CLP mice, but was slightly higher than that in PD-L1WT/WT mice in the sham-operation group and PD-L1flox/flox mice (scale bar, 50 μm) (n = 5). *P < .05, 1-way ANOVA. (F) After CLP, PD-L1flox/flox CLP mice demonstrated higher 7-day survival rates than PD-L1WT/WT CLP mice (n = 8 for PD-L1WT/WT and PD-L1flox/flox mice in the sham-operation group; n = 18 for PD-L1WT/WT and PD-L1flox/flox CLP mice). *P < .05, by log-rank test.
Genetic deletion of neutrophil-specific PD-L1 in vivo decreases proinflammatory plasma cytokine expression, thus limiting lung injury and neutrophil infiltration in the lung and decreasing mortality. (A-C) Plasma concentration of TNF-α (A), IL-6 (B), and IL-10 (C) were determined by enzyme-linked immunosorbent assay. Plasma concentrations of TNF-α and IL-6 were significantly reduced in PD-L1flox/flox CLP mice when compared with levels in PD-L1WT/WT CLP mice, but were still higher than in sham-treated PD-L1WT/WT mice or sham-treated PD-L1flox/flox mice. IL-10 concentration was significantly elevated in PD-L1flox/flox CLP mice when compared with PD-L1WT/WT CLP mice (n = 6). *P < .05, 1-way analysis of variance [ANOVA]. (D) Lung injury score and the representative histopathological images of the lungs. PD-L1flox/flox CLP mice demonstrate significantly lower lung injury score when compared with PD-L1WT/WT sham-treated mice, PD-L1flox/flox sham-treated mice, and PD-L1WT/WT CLP mice (scale bar, 100 μm) (n = 5). *P < .05, Kruskal-Wallis test with Dunn’s multiple comparisons test. (E) Representative immunofluorescence images of MPO (green) staining with blue 4′,6-diamidino-2-phenylindole (DAPI) nuclear stain in lung tissue. The percentage of MPO+ cells was significantly lower in PD-L1flox/flox CLP mice when compared with that in PD-L1WT/WT CLP mice, but was slightly higher than that in PD-L1WT/WT mice in the sham-operation group and PD-L1flox/flox mice (scale bar, 50 μm) (n = 5). *P < .05, 1-way ANOVA. (F) After CLP, PD-L1flox/flox CLP mice demonstrated higher 7-day survival rates than PD-L1WT/WT CLP mice (n = 8 for PD-L1WT/WT and PD-L1flox/flox mice in the sham-operation group; n = 18 for PD-L1WT/WT and PD-L1flox/flox CLP mice). *P < .05, by log-rank test.
In summary, our data suggest that PD-L1 triggers PI3K–dependent prosurvival signaling in neutrophils in clinical and experimental sepsis, but whether PD-L1 is constitutively active in neutrophils remains to be investigated further.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (grants 82072147, 81571935, and 81772105), the Shanghai Pujiang Talent Project (grant 16PJD002), and the Canadian Institutes for Health Research (CHIP; grant, MOP129493).
Authorship
Contribution: J.-f.W., J.C.M., and X.m.D. conceived and designed the study with assistance from S.-h.J. and J.P.; J.-f.W., Y.-p.W., J.X., S.G., S.-h.J., and J.P. performed the in vitro experiments; J.-f.W., Y.-p.W., J.X., Z.-z.Z., and Y.G. performed the in vivo experiments; J.f.W. and X.-m.D. wrote the manuscript; and J.C.M. and X.-m.D. supervised the entire project.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Xiao-ming Deng, Faculty of Anesthesiology, Changhai Hospital, Naval Medical University, 168 Changhai Rd, Shanghai 200433, China; e-mail: deng_x@yahoo.com; and John C. Marshall, Departments of Surgery and Critical Care Medicine, St Michael’s Hospital, University of Toronto, 4-007 Bond Wing, 30 Bond St, Toronto, ON M5B 1W8, Canada; e-mail: john.marshall@unityhealth.to.
Original data can be obtained by e-mail request to either of the corresponding authors.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked ”advertisement” in accordance with 18 USC section 1734.