

In this issue of Blood, Clough et al1 report an induced pluripotent stem cell (iPSC) model of SF3B1-mutant MDS that recapitulates ring sideroblast formation during in vitro erythroid differentiation. Their findings show that coordinated mis-splicing of mitochondrial transporters TMEM14C and ABCB7 by mutant SF3B1 sequesters iron in mitochondria, causing ring sideroblast formation.
In Ancient Greek, σιδηρος (sideros) means iron. In 1954, Kaplan et al used the term “sideroblasts” to define erythroblasts containing cytoplasmic iron granules stainable with the Prussian blue (or Perls) reaction, analogous to the term “siderocytes” that was already in use to designate circulating red cells containing blue-staining granules.2 Using the electron microscope, Bessis and Breton-Gorius found that these granules were aggregates of ferritin.3 In normal individuals, approximately one-third of bone marrow erythroblasts contain 1 to 3 Perls-positive granules in their cytoplasm, representing endosomes filled with excess iron not used for heme synthesis (siderosomes). These normal erythroblasts are defined as “ferritin sideroblasts.”
The term “ringed (or ring) sideroblasts” was coined by Bowman in 1961 to define abnormal sideroblasts that have exceptional accumulations of iron-positive granules surrounding the nucleus like a ring.4 Bowman described 2 categories of patients with ring sideroblasts: (1) cases of refractory normoblastic anemia with ineffective erythropoiesis, and (2) young males with hereditary, hypochromic anemia with hyperplastic, normoblastic marrow.4 These 2 categories are now defined as myelodysplastic syndrome (MDS) with ring sideroblasts (MDS-RS), and X-linked sideroblastic anemia associated with ALAS2 mutations, respectively. Bessis and Breton-Gorius found that in patients with ring sideroblasts, iron granules were not cytoplasmic aggregates of ferritin, but rather perinuclear mitochondria filled with iron-containing material termed “ferruginous micelles.”3 More recently, we showed that the iron deposited in perinuclear mitochondria of ring sideroblasts is present in the form of mitochondrial ferritin, a protein encoded by an intronless gene and highly expressed only in the testis under normal conditions.5
When, as a trainee in hematology, I started examining bone marrow aspirates from patients with refractory anemia, I found ring sideroblasts not only extremely useful for the recognition of morphologic dysplasia but also beautiful. Images like those reported in the figure reminded me of paintings of the French “pointillisme” (see figure). However, despite my strong interest, it took me a long time to understand their pathogenesis in MDS. In 2010, within the Chronic Myeloid Disorders Working Group, we decided to employ massively parallel sequencing technology to identify somatic mutations in MDS patients. We reasoned that ring sideroblasts represented a distinctive morphological abnormality that was likely to be underpinned by distinctive genetic lesion(s). Indeed, of the 8 patients with MDS-RS we initially examined, 6 had a somatic mutation in SF3B1.6 Follow-up studies showed a close relationship between somatically acquired SF3B1 mutation and ring sideroblasts in myeloid neoplasms.7
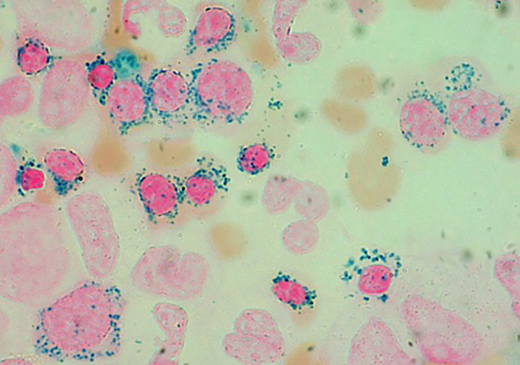
Prussian blue staining of a bone marrow smear from a patient with MDS-RS (Division of Hematology, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy). Ring sideroblasts include both polychromatic and orthochromatic erythroblasts. Original magnification, × 1250.
Prussian blue staining of a bone marrow smear from a patient with MDS-RS (Division of Hematology, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy). Ring sideroblasts include both polychromatic and orthochromatic erythroblasts. Original magnification, × 1250.
Exploiting a unique morphological abnormality was instrumental in defining a new disease paradigm, which opened novel avenues of research. In a subgroup of MDS-RS patients with a somatic SF3B1 mutation, this was the only genetic lesion found. Therefore, we asked a fundamental question: how does a somatic mutation in a splicing factor gene result in a clinical phenotype. Precursor messenger RNA splicing is catalyzed by the spliceosome, a macromolecule composed of small nuclear RNAs associated with proteins, with ∼100 000 spliceosomes in every human cell. As the SF3B1 mutation is heterozygous in MDS-RS, hematopoietic cells contain both normal and SF3B1-mutant spliceosomes, approximately in the same proportions. In turn, this means that about half of splicing events are run by normal spliceosomes, whereas the remaining half are controlled by spliceosomes with a mutant SF3B1 splicing factor. To understand the consequences of this dual RNA splicing, we performed transcriptomic analyses of bone marrow cells from MDS patients.8 SF3B1 mutation was associated with aberrant 3′ splice-site selection, degradation of mis-spliced transcripts because of a premature termination codon (nonsense-mediated decay), and reduced production of canonical transcripts. Three of mis-spliced genes (ABCB7, PPOX, and TMEM14C) were involved with heme biosynthesis and mitochondrial iron metabolism. This suggested that the defective production of the proteins encoded by these genes played a role in ring sideroblast formation, but there was no convincing experimental proof of this hypothesis.9
In their elegant studies, Clough et al show first that iPSC-derived MDS-RS progenitors efficiently form ring sideroblasts in late-stage erythroblasts. Then, using deep RNA-seq, they studied mutant SF3B1 mis-splicing during iPSC erythroid differentiation. Approximately 100 genes were mis-spliced throughout erythroid differentiation, and aberrant transcripts included TMEM14C, PPOX, and ABCB7, resulting in a significant reduction of protein expression. Whereas functional rescue of PPOX had no effect on ring sideroblast formation, that of TMEM14C and ABCB7 markedly reduced this process; of note, combined TMEM14C/ABCB7 rescue nearly abolished ring sideroblast formation. Thus, coordinated mis-splicing of TMEM14C and ABCB7 causes ring sideroblast formation in SF3B1-mutant MDS.
This study clarifies the pathogenesis of the striking morphologic abnormality of patients with refractory normoblastic anemia that Bowman described and named 60 years ago.4 It represents a prototype of the functional studies that should be conducted in splicing factor mutant-neoplasms to understand how abnormal splicing results in abnormal cell differentiation and maturation. Ideally, these studies should be aimed not only at deciphering disease pathogenesis but also at developing novel effective treatments.
SF3B1-mutant MDS is a distinct disease subtype, mainly characterized by ineffective erythropoiesis and a relatively indolent clinical course.10 The major factor in the pathogenesis of anemia in SF3B1-mutant MDS is represented by the apoptosis of late-stage erythroblasts, that is, polychromatic and orthochromatic erythroblasts. As these erythroid precursors are also characterized by ring sideroblast formation, a causal relationship between mitochondrial iron overload and increased propensity to apoptosis is likely. However, this relationship needs to be verified experimentally, and this might be the next task of the Doulatov laboratory in Seattle. Patients with MDS-RS may respond to luspatercept with the abolition of their transfusion requirement, but how this compound targets ineffective erythropoiesis in this MDS subtype has never been definitely documented. Verifying how luspatercept improves red cell production in MDS-RS is therefore important. However, the approach described by Clough et al might also lead to the identification of new targets and the discovery of novel drugs.
Conflict-of-interest disclosure: The author declares no competing financial interests.

