In this Edition of Blood, Pinto et al1 from the WebThal Consortium present an important 10-year follow-up of a cohort of thalassemia patients with invasively documented pulmonary hypertension. The study presents several key findings. Firstly, pulmonary arterial hypertension (PAH) was quite deadly, even with subsystemic pressures, and patients were undertreated by current standards of care. Secondly, change in predicted pulmonary artery pressure by echocardiography was the only significant predictor of survival.
The authors’ initial report documented a 2.1% prevalence of catheterization-documented PAH in thalassemia,2 more than 10 000 times higher than the general population. However, PAH prevalence measured by echocardiogram is at least an order of magnitude higher. There are many reasons for the “false positivity” of echocardiograms, but the differences can be readily corrected by adjusting thresholds for the tricuspid regurgitation jet velocity (TRV) to a more stringent thresholds, such as the 3.2 meters-per-second threshold used in the WebThal study. The downside with being too strict, however, is that it limits diagnosis to advanced disease. In the present report, PAH mortality was 22%, 35%, and 40% at 1, 2, and 5 years following diagnosis. The WebThal study designated TRV values of 3.0 and 3.1 as PAH possible but did not catheterize them or report their clinical outcomes. Given the malignant nature of PAH, it has been my clinical practice to extend right heart catheterization and subsequent therapy (when appropriate) to patients with TRV velocities of ≥3.0. A TRV threshold ≥3.0 lowers the positive predictive value of echocardiogram, but improves detection of potentially reversible PAH.
The second, sad, point highlighted by the current report is that PAH is undertreated in thalassemia, once recognized. In the present report, 1 out of 8 patients received no therapy, and only 29.2% had more than a single agent used. Although the WebThal study was underpowered to demonstrate the advantages of dual therapy, current American College of Chest Physician’s consensus guidelines recommend combination therapy of phosphodiesterase type 5 inhibitor and endothelin receptor agonist as first-line therapy in symptomatic patients.3 Three complementary physiologic axes to treat pulmonary hypertension, the nitric oxide, endothelin, and prostaglandin pathways, create ample opportunity for drug synergy. The endothelin pathway also overlaps the renin-angiotension-aldosterone axis, creating an additional level of synergy. Sequential add-on therapy for pulmonary artery hypertension is also Class 1B recommendation from the European Respiratory Society/European Society of Cardiology. Given the complexity of multidrug therapy, and the rapidly changing treatment landscape, thalassemia patients with PAH should be referred to pulmonary hypertension specialists at tertiary centers rather than being managed by general cardiologists.3
On a more positive note, the recent WebThal study suggests that trends in systolic pulmonary artery pressure (sPAP) predicted by TRV (sPAP = 4 × TRV2) represent a good biomarker to track response to therapy, with 25% improvement in estimated sPAP associated with survival over the study interval.1 This provides a benchmark to determine treatment escalation in thalassemia patients with PAH.
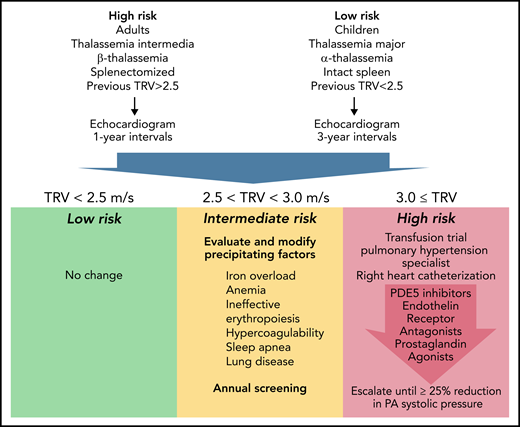
Taking the WebThal reports into account, proposes a management algorithm (see figure). It represents my personal opinion but could serve as a starting point toward consensus guidelines. Echocardiogram screening is recommended for all thalassemia patients, but the screening interval is informed by patient age,2 genotype,4 spleen status,2 transfusion status,5 and prior TRV results.
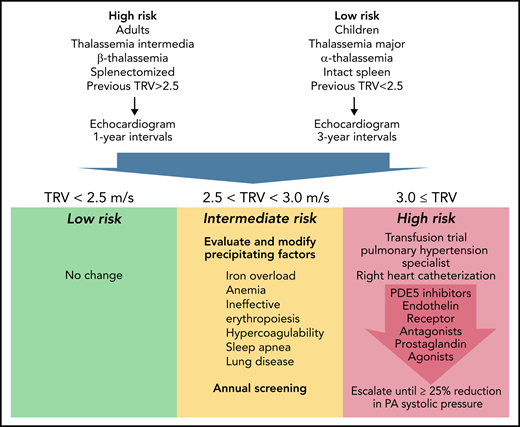
Proposed Algorithm For Screening and Treatment of Pulmonary Hypertension in Thalassemia. Represents candidate pulmonary arterial hypertension guidelines informed by the WebThal data. The risk of pulmonary hypertension increases with age and splenectomy, as well as the severity of anemia and ineffective erythropoiesis (which is impacted by genotype). The frequency of echocardiogram screening should reflect these apriori risks, as well as previous echocardiogram results. TRV results <2.5 meters per second are normal and require no change in monitoring or therapy. TRV results between 2.5 and 2.9 meters per second indicate an abnormal physiology that could potentially progress to pulmonary arterial hypertension. The hematologist must carefully examine potential underlying factors and try to ameliorate them, using serial TRV assessment as the biomarker of success. Regardless of apriori pulmonary hypertension risk, a documented abnormal TRV above 2.5 meters per second suggests a high-risk phenotype and annual follow-up. A TRV of 3.0 meters per second or higher demarcates a patient at significant risk for catheter-proven pulmonary artery hypertension. A first, critical, step is initiation or intensification of chronic transfusion therapy to completely suppress endogenous erythropoiesis. In early disease, this can be sufficient to normalize pulmonary artery pressures. If unsuccessful, the patient should be referred to a pulmonary hypertension specialist for right heart catheterization and aggressive titration of pulmonary hypertension therapies to a target goal of 25% reduction in pulmonary artery systolic pressure.
Proposed Algorithm For Screening and Treatment of Pulmonary Hypertension in Thalassemia. Represents candidate pulmonary arterial hypertension guidelines informed by the WebThal data. The risk of pulmonary hypertension increases with age and splenectomy, as well as the severity of anemia and ineffective erythropoiesis (which is impacted by genotype). The frequency of echocardiogram screening should reflect these apriori risks, as well as previous echocardiogram results. TRV results <2.5 meters per second are normal and require no change in monitoring or therapy. TRV results between 2.5 and 2.9 meters per second indicate an abnormal physiology that could potentially progress to pulmonary arterial hypertension. The hematologist must carefully examine potential underlying factors and try to ameliorate them, using serial TRV assessment as the biomarker of success. Regardless of apriori pulmonary hypertension risk, a documented abnormal TRV above 2.5 meters per second suggests a high-risk phenotype and annual follow-up. A TRV of 3.0 meters per second or higher demarcates a patient at significant risk for catheter-proven pulmonary artery hypertension. A first, critical, step is initiation or intensification of chronic transfusion therapy to completely suppress endogenous erythropoiesis. In early disease, this can be sufficient to normalize pulmonary artery pressures. If unsuccessful, the patient should be referred to a pulmonary hypertension specialist for right heart catheterization and aggressive titration of pulmonary hypertension therapies to a target goal of 25% reduction in pulmonary artery systolic pressure.
TRV <2.5 meters per second is normal, and no changes in management are indicated. A TR velocity of 2.5 to 2.9 meters per second suggests an abnormal physiologic state that is potentially reversible and carries a relatively low probability of meeting criteria for pulmonary hypertension on right heart catheterization. Patients in this zone require considerable hematological input, given the impact of anemia, ineffective erythropoiesis, iron overload, clotting disorders, lung disease, and sleep apnea on pulmonary hypertension.6 Careful detective work is necessary to identify and correct inciting factors, taking care to eliminate contributions from left heart disease, chronic thromboembolism, and lung disease. Hydroxyurea therapy has been associated with lower echocardiography estimates of pulmonary artery pressure in thalassemia intermedia patients.7 Activin-trap drugs or other agents that modify the balance of effective and ineffective erythropoiesis also have the potential to impact the risk of PAH but have not been studied in this regard.
When TR velocity is ≥3, a trial of chronic transfusion therapy (or refinement of existing transfusion therapy) is absolutely warranted. In one thalassemia intermedia patient, we successfully reversed catheter documented PAH by simply shortening transfusion intervals from 6 weeks to 3 weeks (unreported data), using transfusion criteria identical to thalassemia major. If transfusion therapy fails, a pulmonary hypertension specialist should be consulted, right heart catheterization performed, and PAH therapies initiated. In small series, sildenafil has shown efficacy in both short term8 and long-term administration.9 Bosentan and intravenous prostaglandin therapy have been described only in case reports.10 Other agents, such as calcium channel blockers, treprostinil, guanalate cyclase stimulators, and nebulized iloprost, have never been reported in thalassemia but are routinely used in other forms of pulmonary artery hypertension.6 The WebThal data presented by Pinto et al suggest that therapeutic response to treatment was more predictive of survival than initial pulmonary artery pressure itself. Therefore, I recommend that therapies should be uptitrated with the goal of lowering predicted pulmonary artery systolic by at least 25%, using multiple agents if necessary to accomplish this.
Where next for this awful complication? From a basic science perspective, many gaps remain between observed clinical risk factors and pathophysiological mechanisms. Elucidation of the basic mechanisms is likely to inform therapies. With respect to patient management, the heterogeneity of patient phenotypes, transfusion, splenectomy practices, and monitoring across thalassemia centers makes it challenging to compare studies of pulmonary hypertension prevalence and management. Consortia studies and patient registries represent the only cost-effective mechanisms to study clinical outcomes. Individually, hematologists must take responsibility to screen their thalassemia patients appropriately, diligently manage the risk factors under their control, and refer patients at highest risk to pulmonary hypertension specialists. Lastly, cardiologists and hematologists experienced with thalassemia need to strive toward consensus, rather than ad-hoc, management guidelines.
Conflict-of-interest disclosure: J.C.W. consults for Celgene, WorldCareClinical, ImagoBiosciences, and Silence Therapeutics. He receives research support from the National Institutes of Health (NIH), Philips Medical Systems, the Saban Research Institute, and the Additional Ventures Single Ventricle Fund.