Abstract
Platelets (small, anucleate cell fragments) derive from large precursor cells, megakaryocytes (MKs), that reside in the bone marrow. MKs emerge from hematopoietic stem cells in a complex differentiation process that involves cytoplasmic maturation, including the formation of the demarcation membrane system, and polyploidization. The main function of MKs is the generation of platelets, which predominantly occurs through the release of long, microtubule-rich proplatelets into vessel sinusoids. However, the idea of a 1-dimensional role of MKs as platelet precursors is currently being questioned because of advances in high-resolution microscopy and single-cell omics. On the one hand, recent findings suggest that proplatelet formation from bone marrow–derived MKs is not the only mechanism of platelet production, but that it may also occur through budding of the plasma membrane and in distant organs such as lung or liver. On the other hand, novel evidence suggests that MKs not only maintain physiological platelet levels but further contribute to bone marrow homeostasis through the release of extracellular vesicles or cytokines, such as transforming growth factor β1 or platelet factor 4. The notion of multitasking MKs was reinforced in recent studies by using single-cell RNA sequencing approaches on MKs derived from adult and fetal bone marrow and lungs, leading to the identification of different MK subsets that appeared to exhibit immunomodulatory or secretory roles. In the following article, novel insights into the mechanisms leading to proplatelet formation in vitro and in vivo will be reviewed and the hypothesis of MKs as immunoregulatory cells will be critically discussed.
Introduction
Megakaryocytes (MKs) are derived from hematopoietic stem cells (HSCs), which, because of their life-long self-renewal capacity, can give rise to a plethora of progenitor cells with increasing lineage specificity.1 MK differentiation is induced upon interaction of thrombopoietin (TPO) with its receptor myeloproliferative leukemia protein (c-Mpl) on the HSC surface, which stimulates an intracellular signaling cascade involving Janus kinase 2 and the transcription factors signal transducers and activators of transcription 3 and 5 (STAT3 and -5). In addition to STAT proteins, which translocate to the nucleus and induce transcription of MK-specific gene signatures including the temporal upregulation of several transcription factors, such as GATA1 and -2, Fli1, and RUNX1.2 TPO signaling further induces activation of protein kinase C and phosphoinositide 3-kinase.3-6 Deficiency in either TPO or its receptor c-Mpl leads to marked thrombocytopenia related to abolished MK maturation and further affects the abundance of long-term progenitors thus implying an important role of the receptor in maintaining HSC proliferation.7 However, the observation that a low number (∼10%) of platelets is still maintained after TPO signaling is abolished indicates other signaling mechanisms by which MK maturation and subsequent platelet production can be induced, among them signaling through interleukin 1α (IL1α), C-C motif chemokine ligand 5 (CCL5), insulin-like growth factor 1 (IGF1), and iron.8-11 A recent publication further elucidated that absent CXCL4/platelet factor 4 (Pf4) secretion from MKs rather than TPO itself is responsible for impaired HSC quiescence in TPO-deficient mice.12 The distinct activation of specific transcription factors is essential for MK differentiation, which is characterized by cytoplasmic maturation including the formation of an elaborate membrane system (the demarcation membrane system [DMS]) and polyploidization without cytokinesis, the so-called endomitosis leading to ploidy levels ranging from 4 to 128N. The main function of mature MKs is the generation of platelets; however, several recent studies identified additional subsets of MKs that serve as immunomodulatory cells in both bone marrow (BM) and distant organs, such as lung and liver.13-15 This review was conducted to inform on the latest advances in understanding MK heterogeneity and highlights our current knowledge on proplatelet formation dynamics in vitro and in vivo.
Making their nest: MKs within the BM microenvironment
Megakaryopoiesis has long since been described to follow hierarchical structures, with HSCs giving rise to multipotent progenitors that emerge into MK/erythroid progenitors, which then further differentiate into MK progenitors and ultimately into mature MKs.16 However, this hierarchical model of MK differentiation was challenged by the observations of lineage-restricted multipotent progenitors with self-renewal capacity and heterogeneity within the HSC subpopulations, which was supported by single-cell omics analyses.14,17,18 MKs can easily be distinguished from other cells, not only by their cell size (ranging from 20-100 µm), but also because of the expression of cell-specific glycoproteins (eg, glycoprotein [GP] VI, integrin αIIbβ3) and soluble mediators, such as Pf4 and von Willebrand factor (vWF), which notably is also highly expressed in endothelial cells.19 Despite an overlap in the expression of a variety of receptors between MKs and HSCs, among them CD150, CXC receptor 4 (CXCR4), and c-Mpl,20 only the recent discovery of vWF+ HSCs, localized in proximity to mature MKs, gave insights into a nonhierarchical myeloid differentiation,21 which was reinforced in several independent studies revealing an increased abundance of MK-biased hematopoietic progenitors during inflammation,22 myeloproliferative neoplasms,23 and aging,24 thus implying that these progenitors are an alternative pathway for “emergency” megakaryopoiesis. The hypothesis of HSC subsets was supported by the observation of different HSC-sustaining niches within the BM, in which distinct HSC subsets were detected in proximity to MKs and BM arterioles, respectively, thus suggesting disparities in their differentiation potential.21,25 The close contact between HSCs and MKs in the BM has long been said to be essential for maintaining HSC niche capacities and appears to be important for the paracrine secretion of a variety of cytokines and growth factors. Among these, MK-derived Pf4 and TGF-β1 were described as contributing to HSC quiescence, the loss of which resulted in increased HSC proliferation.26,27
Platelet production requires MKs to localize to blood vessel sinusoids. It has been suggested that migration of MKs from the osteoblastic to the vascular niche is therefore an essential process in thrombopoiesis.28,29 The cytokine stromal-derived factor 1 (SDF1) expressed in the vascular niche is thought to drive this migration by interaction with its receptor CXCR4 on MKs.30 In line with this hypothesis, expression of SDF1 and fibroblast growth factor 4 (FGF4) restored thrombopoiesis in thrombocytopenic mice lacking TPO or its receptor c-Mpl.28 The expression and/or activity of CXCR431 and c-Mpl32 on MKs is regulated by dynamins, and inhibition of these GTPases resulted in impaired receptor endocytosis and enhanced activity of the cytoskeletal regulator RhoA that has been implied in MK transmigration into vessel sinusoids.33 Accordingly, it has been suggested that MK migration toward the vascular niche is subject to cytoskeletal rearrangements. In line with this notion, patients with mutations within MYH9, the gene encoding nonmuscle myosin IIA (NMIIA), which was recently shown to mediate MK migration, display coagulation defects attributable to thrombocytopenia.34,35 It has been proposed that loss or gain of contractile function of NMIIA or misalignment of the actomyosin cytoskeleton and subsequent aberrant NMIIA function, are causative of the mislocalization of MKs within the BM in corresponding mouse models.36 Recently, the hypothesis of MK migration was challenged in consecutive studies in which a light-sheet microscopy approach was used combined with mathematical simulations.37-39 The observation of very slow MK migration, limited to the vessel-biased MK pool, suggests that MKs at the sinusoids are replenished by precursor cells rather than migrating from the osteoblastic niche. In addition to direct cell interactions and cytokine gradients, MK maturation, their potential migration and platelet release are highly dependent on extracellular matrix (ECM) proteins. Collagen type 1 was identified as an inhibitor of MK maturation and proplatelet formation by increasing ECM stiffness40 and concomitantly interfering in MK signaling pathways through GPVI activation.41 Increased ECM stiffness is also thought to contribute to impaired platelet production in a variety of myeloproliferative neoplasms, in which BM matrix stiffness is markedly enhanced by impaired matrix degradation and pathologically increased production.42,43 It is anticipated that further experiments will delineate whether MK migration is a phenomenon occurring in the BM of humans and mice, how the process might be targeted therapeutically, and whether it is restricted to certain subsets of MKs, which are further defined herein.
Not all MKs are created equal
MKs demonstrate clear cellular heterogeneity, with one of the most pronounced features being variations in nuclear ploidy.44,45 In recent years single-cell RNA sequencing (scRNA-seq) has been a powerful tool for exploring cellular heterogeneity in a broad array of cell types.46-48 In an early study, Davizon-Castillo et al identified variabilities in transcriptomics within native BM MKs; however, these were attributed to reflect different maturation stages of MKs rather than cellular heterogeneity.49 More recent scRNA-seq endeavors on BM MKs have now revealed transcriptional heterogeneity within different MK subsets and have led to the identification of 4 to 5 distinct MK subpopulations in both humans and mice (Figure 1).14,50,51 One of these subsets (Figure 1; subset A) is found to be enriched for genes associated with proplatelet formation (eg, Tubb1 and Myh9) and platelet function (eg, Vwf and Gp1ba), leading to the hypothesis that this subset represents MKs specifically dedicated to thrombopoiesis. In situ staining for the main discriminatory marker for this subset, aryl hydrocarbon receptor nuclear translocator–like protein 1 (Arntl1), verified that MKs were indeed in proximity to sinusoidal blood vessels and were, at least ex vivo, producing proplatelets.14
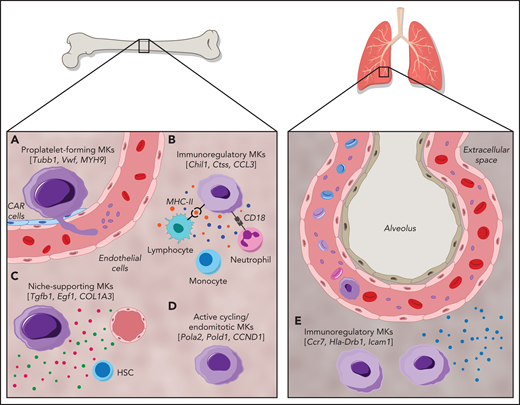
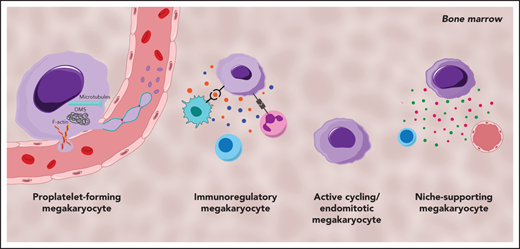
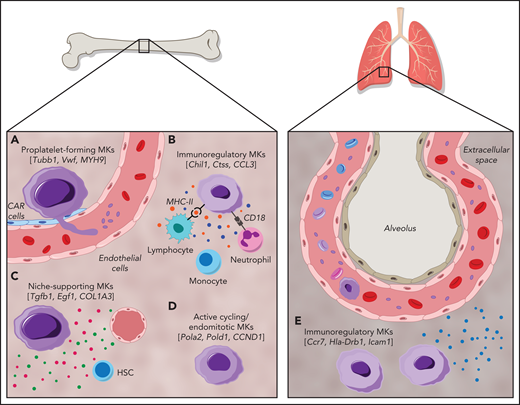
Cellular heterogeneity of MKs within the BM and of MKs present in the lung. MK subgroups within the BM. (A) Proplatelet-forming MKs close to the sinuous blood vessels are high in expression of Tubb1, Vwf,14 and MYH9.51 (B) Immunoregulatory MK subpopulation presenting MHC-II53 and CD18,58 which are responsible for the interaction with lymphocytes and neutrophils, respectively. Immunoregulatory MKs are high in expression of Chil1, Ctss,14 and CCL3,51 and secrete cytokines. (C) MKs, with suggested hematopoietic stem cell niche and osteogenic niche supporting a role evident in the high expression of Tgfb1, Egf1, COL1A3.51 (D) Subset of actively cycling MKs expressing Pola2, Pold1,14 and CCND1.50 (E) The lung contains MKs with an immunoregulatory phenotype expressing Ccr7, Hla-Drb1, and Icam1,13 in addition to intravascular MKs.69
Cellular heterogeneity of MKs within the BM and of MKs present in the lung. MK subgroups within the BM. (A) Proplatelet-forming MKs close to the sinuous blood vessels are high in expression of Tubb1, Vwf,14 and MYH9.51 (B) Immunoregulatory MK subpopulation presenting MHC-II53 and CD18,58 which are responsible for the interaction with lymphocytes and neutrophils, respectively. Immunoregulatory MKs are high in expression of Chil1, Ctss,14 and CCL3,51 and secrete cytokines. (C) MKs, with suggested hematopoietic stem cell niche and osteogenic niche supporting a role evident in the high expression of Tgfb1, Egf1, COL1A3.51 (D) Subset of actively cycling MKs expressing Pola2, Pold1,14 and CCND1.50 (E) The lung contains MKs with an immunoregulatory phenotype expressing Ccr7, Hla-Drb1, and Icam1,13 in addition to intravascular MKs.69
A second subset (Figure 1; subset B) of BM MKs with a low ploidy (2-4N) has transcription profiles enriched for inflammatory markers such as chitinase 3–like 1 and 3 (Chil1 and Chil3) and the tetraspanin CD53, as well as genes involved in cytokine production and release, indicating a specific subgroup of immunoregulatory MKs.14 An independent scRNA-seq study on human BM MKs also identified a subset of cells with an immune profile, positive for the lymphocyte activation molecule CD48.50 Systemic infection with Escherichia coli in mice markedly increased the abundance of these CD48+ MKs in the BM, thus substantiating the hypothesis of an active contribution of MKs during immune challenges, although the exact mechanisms of their participation in immune surveillance remains elusive.52 Evidence of an immunoregulatory role of MKs was also presented before the scRNA-seq area, where it was shown that MKs express major histocompatibility complex class 1 (MHC1) molecules and thereby activate T cells through antigen presentation, a phenomenon that increased greatly during inflammatory events, such as sepsis.53,54 In addition to MHC1, MKs express a plethora of other immune cell markers such as FcgRIIA (CD32A) and several members of the Toll-like receptor family that recognize microbial antigens.55,56 Interestingly, MKs were also shown to express CD40L, the ligand for the lymphocyte receptor CD40, which would thus facilitate an interaction of MKs with B and T cells.57 However, how BM MKs would come into contact with mature lymphocytes as proposed in the latest study by Liu et al remains to be determined, because the BM is mainly populated by lymphoid progenitors.50 The hypothesis of interactions between MKs and immune cells was substantiated by previous studies that observed the internalization of immune cells (mainly neutrophils) by MKs in a process referred to as emperipolesis.58 The process was observed under physiological conditions,59 but markedly increased under disease settings such as myelofibrosis.60 MKs were further shown to produce extracellular vesicles (EVs), and it has been hypothesized that MK EVs may encompass immunoregulatory molecules, given that platelet EVs were recently shown to potentiate BM inflammation.61,62 The finding of an immunoregulatory MK subset is thus consistent with the hypothesis of a role for MKs in innate and, more importantly, adaptive immunity.
A third subset (Figure 1; subset C) of murine BM MKs identified by scRNA-seq were enriched for Fgf9 and Fgf10, cytokines that support osteoclast and fibroblast differentiation, respectively.14 In addition to the HSC niche-supporting cytokines IGF1 and PF4,26,63 a comparable subset of human BM MKs was highly positive for collagen type 1 α1 chain and collagen type 3 α1 chain, which are similarly essential for osteoblast maintenance.64 Together, these findings strongly support the notion of a role for MKs in osteoblast and HSC niche maintenance. In contrast, the fourth MK subgroup (Figure 1; subset D) comprised low-ploidy, actively cycling cells, which was evident by the high expression of DNA polymerase α subunit 2 and DNA polymerase δ subunit 2, both of which encode for subunits of the DNA polymerase complex. Although the investigators hypothesized that this actively cycling MK population can give rise to all 3 previously described subsets,14 this idea should be subjected to further investigation.
Megakaryopoiesis is a complex process starting with the development of the hematopoietic system in the third week of human development (embryonic day 7.5 in mice) and takes place in the fetal liver and yolk sac.65,66 The heterogeneity visible among adult BM MKs is similarly present in MKs derived from yolk sacs and fetal livers of human embryos.51 Interestingly, both tissues contain an additional MK subset, which highly expresses serine peptidase inhibitor kazal type 2 (SPINK2) and CD7, which the researchers allocated to an immature MK subpopulation. This fifth subset was also demonstrated by another independent study on human BM MKs that found a subset of MKs enriched in cyclin D1 and actin depolymerizing factor, genes necessary for endomitosis.50 One could hypothesize that cellular heterogeneity is a representation of MKs at different stages of differentiation rather than distinctive subsets. This notion is contradicted, however, by pseudotime analyses of yolk sac and fetal liver MKs, where thrombopoietic and immunoregulatory MKs are generated along 2 distinct developmental routes.51 Overall, scRNA-seq has provided novel insights into heterogeneity among MKs and substantiated the notion of MK functions beyond platelet production. It must be noted, however, that functional validation of these subpopulations is so far limited and remains territory for further invesitgation.14,50,51 In addition, despite first attempts using in situ immunostaining, the challenge that remains is to fully capture the spatial orientation of these MK subpopulations within the BM and other organs.
MKs beyond the borders of the BM
The first report of MKs residing in distant organs such as the lung dates back to 1893.67 Analysis of MKs in the blood of human subjects with a cardiac catheter supported this idea: MK levels were lower in blood leaving the lung vs blood directed toward the lung.68 It was not until recently, however, that the function of lung-resident MKs gained more attention. Two-photon intravital microscopy of murine lungs showed that MKs circulate through the lung and dynamically release platelets into the lung vasculature (Figure 1).69 Initial studies suggested variable contributions of lung MKs to the circulating platelet pool, with percentages of 7% to 50% reported.69,70 This latter percentage has been a topic of debate, because the data are solely based on extrapolations of single events observed in lung tissue and therefore need to be corroborated. Moreover, if the lung was a major site of platelet production, one would expect to see thrombocytopenia in patients with lung disease, which is generally not observed. In a separate publication, it was emphasized that most of the MKs in the lung reside in the interstitial space, suggesting an alternative function of lung MKs, independent of platelet generation.13 Importantly, the transcription profile of lung MKs skews toward a role in immunity and inflammation rather than platelet generation.15 This finding was corroborated by studies in which scRNA-seq revealed that the transcriptional profile of lung MKs closely resembles the immunoregulatory subset identified within the BM (Figure 1).14 Recently, it was also shown that lung MKs exhibited a high expression of immune-related molecules, such as the HLADR isotype and intercellular adhesion molecule 1.13 When subjected to an immune stimulus, such as interferon γ or lipopolysaccharide, expression of these receptors was strongly upregulated indicating an active role of lung MKs in the response to inflammatory stimuli. However, the transcriptome profile of lung-resident MKs also closely resembles dendritic cells, whereas BM MKs exhibited a monocytic expression profile.14 One might therefore question the classification of these cells as MKs rather than immune cells expressing MK markers. Interestingly, patients suffering from the novel coronavirus disease 2019 (COVID-19) displayed increased MKs in lung capillaries.71 Moreover, in a multiomics study, Bernardes et al found a significantly increased number of interferon-activated MKs in whole blood of patients with COVID-19, thus implying their role in immune defense.72 Immunohistology studies performed on the brains of those patients further identified CD61+ cells, presumably MKs, in brain capillaries, where they are thought to contribute to neurological impairments by occluding brain vessels.73 However, because CD61 is expressed on a variety of other cells and the authors provide no further mechanistic insight into their contribution to disease progression, the significance of this observation requires further proof.
How do MKs break through to the other side?
Although the identification of novel MK subsets and their presence in distant organs suggest novel functions of MKs, independent of platelet production, one longstanding question in the field remains how proplatelet-forming MKs (subset A) penetrate the endothelial barrier to deposit platelets into the blood. First, observations of proplatelet-forming MKs identified filamentous actin (F-actin)–rich protrusions reaching through the endothelial barrier,74 which were later classified as podosomes. Intravital imaging of cranial BM revealed polarization and protrusion of MKs through the sinusoidal barrier, which ultimately allowed for platelet release into the bloodstream.75 Podosomes consist of an actin-rich core, interconnected by actin-related proteins (Arp2/3), which in turn is surrounded by adhesion molecules, such as talin and vinculin, and were first described to occur in migrating cell types with invasion capacity, such as macrophages and osteoclasts.76 Subsequent studies confirmed their presence in MKs77 and identified matrix metalloproteinase secretion through podosomes to markedly contribute to the degradation of basement membrane proteins, thus facilitating protrusions through the endothelium into vessel sinusoids.78 Eckly et al showed that formation of Arp2/3-dependent podosomes is responsible for endothelial breaching during the initiation of proplatelet formation (Figure 2).79 Their findings, in line with previous data,80 suggest that MKs form large protrusions through transendothelial pores, which are indispensable to enable the release of proplatelets into the circulation. This effect is supported by findings in mice lacking the Rho GTPases Rac1 and/or Cdc42 that present with severe thrombocytopenia related to reduced podosome formation.81,82 Of note, in addition to significantly altered actin dynamics, mice deficient for Rac1 and Cdc42 exhibited defective tubulin reorganization, thus emphasizing how both cytoskeletal components interact to enable proplatelet formation. Another recent publication postulated that protrusion-forming MKs colocalize with CXCL12-abundant reticular cells, which are closely associated with both sinusoidal endothelium and the basal lamina, thus supporting the hypothesis that MK-endothelial/reticular cell cross talk induces initial protrusion formation (Figure 2).83

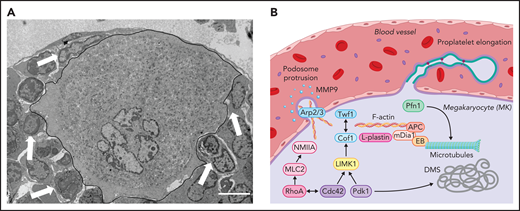
Intracellular mechanisms of proplatelet formation and elongation. (A) Transmission electron micrograph of a BM MK in contact with an endothelial cell. MK is outlined in black. Arrowspoint towards other BM cells interacting with the MK. Scale bar, 3 µm. (B) Platelet production from MKs is enabled through extensive cytoskeletal rearrangements leading to breaching of the endothelium through the formation of podosomes and proplatelet generation, during which proplatelets appear like “beads on a string” and function as essential intermediate structures. Initial polarization of the DMS is dependent on actin dynamics enabled by the Rho GTPase Cdc42, which induces LIMK1 activity and activates downstream effectors such as Cof1 and Twf1 and is further indispensable to enable proplatelet formation.81,82,99 In addition to Cdc42 and PDK1,102 activity of another Rho GTPase, RhoA, which induces MLC2/NMIIA activation,100,101 is critical in enabling F-actin rearrangements including the Arp2/3-dependent formation of podosomes, which are crucial to breach the endothelium, most likely though the secretion of MMPs.78,79 Proplatelet elongation on the other hand relies on microtubule sliding within the proplatelet shaft,117,127 which is dependent on the motor protein dynein.128 Cytoskeletal cross talk is enabled through proteins linking the F-actin to the microtubule cytoskeleton, such as EBs, APC, and mDia1.95,97
Intracellular mechanisms of proplatelet formation and elongation. (A) Transmission electron micrograph of a BM MK in contact with an endothelial cell. MK is outlined in black. Arrowspoint towards other BM cells interacting with the MK. Scale bar, 3 µm. (B) Platelet production from MKs is enabled through extensive cytoskeletal rearrangements leading to breaching of the endothelium through the formation of podosomes and proplatelet generation, during which proplatelets appear like “beads on a string” and function as essential intermediate structures. Initial polarization of the DMS is dependent on actin dynamics enabled by the Rho GTPase Cdc42, which induces LIMK1 activity and activates downstream effectors such as Cof1 and Twf1 and is further indispensable to enable proplatelet formation.81,82,99 In addition to Cdc42 and PDK1,102 activity of another Rho GTPase, RhoA, which induces MLC2/NMIIA activation,100,101 is critical in enabling F-actin rearrangements including the Arp2/3-dependent formation of podosomes, which are crucial to breach the endothelium, most likely though the secretion of MMPs.78,79 Proplatelet elongation on the other hand relies on microtubule sliding within the proplatelet shaft,117,127 which is dependent on the motor protein dynein.128 Cytoskeletal cross talk is enabled through proteins linking the F-actin to the microtubule cytoskeleton, such as EBs, APC, and mDia1.95,97
The process by which initial protrusions transition into microtubule-rich proplatelet shafts, from which platelets are released into the circulation, remains elusive so far. A recent publication challenged the idea of classic proplatelet formation and suggested that membrane budding contributes to the majority of circulating platelets.84 Using novel 2- and 3-dimensional imaging techniques, Potts et al84 provided evidence of the occurrence of membrane budding from fetal liver and BM MKs in vivo. However, they fell short in recognizing the presence of MK-derived EVs formed via blebbing, which are morphologically very similar to “budded” platelets,61,85 and in reviewing the numerous publications visualizing proplatelet release in vivo using 2-photon intravital microscopy of the cranium.37,75,80 Interestingly, the hypothesis of differences between in vitro and in vivo proplatelet formation was also conveyed in another publication, which identified unique cytoskeletal components to account for these disparities.86 Nonetheless and in contrast to the study by Potts et al,84 Bornert et al86 concluded that in vivo proplatelet formation occurs through the release of proplatelet shafts, not through membrane budding.
The MK cytoskeleton as an engine for platelet generation
Even though the contribution of podosome formation and subsequent membrane budding is still controversially discussed, it is widely accepted that protrusion through the basement membrane and the subsequent release of fragile proplatelet shafts into the bloodstream requires constant and fine reorganizations of the 3 major types of cytoskeletal proteins: the actin cytoskeleton, microtubules, and intermediate filaments, all of which are interconnected by integrator proteins such as spectrins or septins. Actin is among the most highly expressed proteins in MKs and platelets with concentrations ranging between 50 to 200 µM.87 Only adenosine triphosphate (ATP)–bound globular (G)-actin monomers can be added to the growing filament, because ATP is hydrolyzed during polymerization. F-actin assembly is mainly regulated by small actin monomer binding proteins. Two actin sequestering proteins compete for the binding to ADP-bound G-actin at the barbed end, thymosin β4 (Tβ4), and profilin 1 (Pfn1).88 Although Pfn1 catalyzes the nucleotide exchange on G-actin, thus promoting F-actin assembly, Tβ4 mainly inhibits addition of actin monomers by reducing the pool of freely available G-actin.89 MK- and platelet-specific loss of Pfn1 in mice results in microthrombocytopenia, ectopic release of platelet-like particles into the BM compartment and hyperstability of microtubules, similar to observations in patients with Wiskott-Aldrich syndrome, a rare bleeding diathesis associated with reduced platelet counts and defective platelet function.90,91 These findings highlight how a functional crosstalk between different cytoskeletal components is essential for proplatelet release and platelet function. The actin-severing proteins cofilin 1 (Cof1) and its cofactor ADF bind to the pointed or (−)end of the actin filament upon ATP hydrolysis, which eventually leads to the dissociation of ADP-bound monomers from the filament.92 Loss of both proteins leads to macrothrombocytopenia in mice, because proplatelet formation is virtually abolished.93 Another group of ADF homology domain–containing proteins, twinfilins (Twf), has been shown to consolidate actin and microtubule cytoskeletal dynamics, together with Cof1, not only by interfering with F-actin depolymerization, but by affecting the expression of the actin nucleating proteins mDia1 and adenomatous polyposis coli (APC).94 Both proteins can interact with the microtubule-associated protein end binding protein 1 (EB1) and thus affect microtubule stability, which is indispensable for a functional release of platelets into the bloodstream.95 Accordingly, deficiency in either mDia1 or APC in human or murine MKs results in increased proplatelet formation due to altered F-actin and microtubule dynamics.96,97 By taking advantage of micro-RNA profiling, an independent study further identified downregulation of the actin bundling protein l-plastin to be critically involved in enabling proplatelet formation, thus shedding light on novel regulators of actin binding proteins in maturing MKs.98 The activity of small-actin monomer binding proteins can be modified by a family of rho GTPases that cycle between an active, guanosine triphosphate–bound, and an inactive, guanosine diphosphate–bound, state. Although RhoA has been said to affect the activity of mDia1 and myosin light chain (MLC) phosphorylation through activation of rho-related kinases, its homologue Cdc42 is indispensable for DMS maturation and controls LIM kinase 1 activity, which in turn phosphorylates and thus deactivates Cof1 (Figure 2).99-101 LIM kinase 1 activity is further controlled by phosphoinositide-dependent kinase 1 (PDK1), a target of phosphoinositide 3-kinase, the loss of which in mice resulted in severe macrothrombocytopenia related to reduced Cof1 deactivation and subsequent impaired F-actin dynamics and DMS formation.102 Lack of Cdc42 or its downstream effectors has also been associated with a compromised DMS development, which is in line with previous observations that identified an essential role for a GPIb/F-actin axis in initiating DMS formation, thus highlighting the essential role of F-actin dynamics in DMS elaboration.103 Interestingly, impaired MK ultrastructural development was observed in a plethora of other mouse models lacking actin-regulatory proteins,93,94,99,104 thus implying that their defects in proplatelet formation could arise from a generally affected cytoskeletal maturation (including a reduced DMS area), rather than a direct involvement of the actin cytoskeleton in proplatelet initiation. The means by which altered actin dynamics in the final stages of proplatelet formation influence platelet release thus remains to be elucidated. Both RhoA and Cdc42 have also been shown to be essential for MK maturation, because MKs deficient in both proteins displayed marked differentiation defects, albeit unaltered ploidy.81 However, RhoA localizes to the cleavage furrow in MKs and a tight regulation of RhoA activity is indispensable to initiate the first endomitotic cycle,105,106 thus suggesting that its deletion in maturing MKs using the Pf4-Cre system occurs at a later stage. The markedly impaired proplatelet formation observed in double-deficient MKs could partly be attributed to a reduced expression of the rho-kinase effector MLC2 and its target NMIIA, the loss of which has been shown to be associated with defective MK maturation and proplatelet dynamics.107 These findings, in conjunction with the thrombocytopenia observed in patients with variants within MYH9,108 highlight the importance of functional F-actin dynamics during MK maturation. This observation is in line with the similarly defective MK maturation and proplatelet formation associated with macrothrombocytopenia in patients with variants in DIAPH1, TPM4, ACTN1, or FLNA, other regulatory proteins essential for F-actin dynamics.109-112
Although the role of the actin cytoskeleton in proplatelet initiation is still controversially discussed, the importance of microtubule reorganization during proplatelet elongation have been clearly demonstrated.113,114 Microtubules consist of α- and β-tubulin heterodimers that can polymerize into protofilaments, 13 of which can form hollow tubes with a fast polymerizing plus (+)end and a slow polymerizing minus (−)end with exposed GDP-bound α-tubulin.115 Microtubules are in a constant state of dynamic instability, which can lead to sudden disassembly of the filament, termed “catastrophe” that can be controlled by microtubule-associated proteins. Among these, EBs can accelerate microtubule polymerization, most likely by modulating the (+)end conformation,116 and enable cross talk of microtubules with the F-actin cytoskeleton.95 The kinesin family of microtubule depolymerases removes terminal tubulin subunits from the filaments through ATP hydrolysis and is related to their function as motor proteins able to move along the filament without dissociation. Another motor protein, the (−)end-directed multiprotein complex dynein, can exert pulling forces onto microtubules, thus inducing the sliding of filaments against one another (Figure 1B).117 Patients with variants within TUBB1, the gene encoding for the most prevalent tubulin isoform in MKs, β1, exhibit pronounced macrothrombocytopenia, which was recapitulated in mice lacking one of its upstream regulators, the transcription factor NF-E2.118,119 Microtubule stability and disassembly rates are mainly coordinated through posttranslational modifications of different tubulin isoforms.120 Both detyrosination and acetylation on α-tubulin have been associated with enhanced stability and/or longevity of the filament attributable to altered binding of EBs,120-122 which was further supported by a study demonstrating that altered microtubule tyrosination in mice lacking the tubulin isoform α4 in MKs accounts for macrothrombocytopenia.123 The spatial regulation of polymodifications, most importantly glutaminylation and glycosylation, on β1-tubulin, however, was recently shown to be dependent on tubulin tyrosine ligase–like molecules and linked to functional proplatelet formation, alteration of which results in macrothrombocytopenia related to impaired platelet generation in humans.124,125 Although recent advances have led to the identification of several molecules responsible for microtubule posttranslational modifications in neurons,126 the mechanism that regulates microtubule durability in MKs remains largely elusive.
Early studies have identified that inhibition of dynein binding to MKs markedly impaired proplatelet dynamics, whereas inhibition of microtubule assembly using the stabilizing drug nocodazole did not affect proplatelet extension rates in vitro.127 Using fluorescent live-imaging microscopy, further analyses revealed that dynein-dependent microtubule sliding rather than directed polymerization at (+)ends was indispensable for proplatelet elongation,128 which is enabled because of the re-entry of looped microtubule bundles into the proplatelet shaft allowing for dynein binding to the neighboring protofilament. In addition to their essential role during proplatelet elongation, microtubules are further critical for the transport of organelles and granules into nascent platelets.129 To equip platelets with a variety of granules that enable them to fulfill their various functions in hemostasis, inflammation, and angiogenesis, granules and other organelles colocalize with the motor protein kinesin, which enables their directed transport into the proplatelet tip.130
Perspectives
Platelet production by MKs has been intensely studied in vitro since the discovery of TPO and its receptor in 1994,131,132 and a variety of signaling molecules and cytoskeletal regulators have been identified to affect MK maturation and platelet production. However, the intracellular cues initiating proplatelet formation, the factors promoting endothelial barrier protrusion, and the drivers of proplatelet release remain a matter of debate that can be elucidated by further study. Although MKs are mainly responsible for maintaining physiological platelet counts, a variety of distinctive MK subsets have recently been identified as fulfilling different roles in immunity and inflammation, not only in the BM but also in distant organs such as the lung or the fetal liver.15 Although initial steps toward understanding these novel MK subsets have been taken, it remains largely unknown if these MKs contribute to innate and adaptive immunity. We hope future studies will aim at delineating which intracellular or extrinsic factors influence MK fate decisions and whether the immunomodulatory MK subset can possibly be targeted to manipulate immune responses upon infection.
Acknowledgments
The authors thank Kristin Johnson for generating the figures in this review, Harvey G. Roweth for commenting on the manuscript, and Anjana Ray for the processing of the samples displayed in Figure 1.
This work was supported by Rubicon grant 452020206 from The Netherlands Organization for Health Research and Development (J.T.) and National Institutes of Health, National Heart, Lung, and Blood Institute grant R01HL68130 (J.E.I.).
Authorship
Contribution: J.T., I.C.B., and J.E.I. designed and wrote the manuscript.
Conflict-of-interest disclosure: J.E.I. has financial interest in and is a founder of PlateletBio, a biotechnology company focused on making donor-independent platelet-like cells at scale. The interests of J.E.I. are managed by Boston Children’s Hospital. J.T. and I.C.B. declare no competing financial interests.
Correspondence: Joseph E. Italiano, Vascular Biology Program, Boston Children’s Hospital, 1 Blackfan Circle, Boston, MA 02115; e-mail: joseph.italiano@childrens.harvard.edu.