Abstract
The inherited thrombocytopenia syndromes are a group of disorders characterized primarily by quantitative defects in platelet number, though with a variety demonstrating qualitative defects and/or extrahematopoietic findings. Through collaborative international efforts applying next-generation sequencing approaches, the list of genetic syndromes that cause thrombocytopenia has expanded significantly in recent years, now with over 40 genes implicated. In this review, we focus on what is known about the genetic etiology of inherited thrombocytopenia syndromes and how the field has worked to validate new genetic discoveries. We highlight the important role for the clinician in identifying a germline genetic diagnosis and strategies for identifying novel causes through research-based endeavors.
Introduction
Megakaryopoiesis and thrombopoiesis are tightly regulated components of hematopoiesis that result in the production and release of up to 1011 platelets daily to maintain a normal concentration of 150 to 400 × 109/L circulating platelets. These cells are required for adequate hemostasis through the formation of a stable clot at sites of blood vessel injury. Thrombocytopenia, traditionally defined as a platelet count of <150 × 109/L, has many causes including immune destruction, medication-induced aplastic anemia, or as a manifestation of an inherited bone marrow failure syndrome. In this review, we focus on the diagnosis and pathogenesis of inherited thrombocytopenias, with a special emphasis on genetics. These diseases represent a growing collection of germline variant–associated thrombocytopenias whose primary manifestation is inadequate circulating platelet numbers. Many of these syndromes have extrahematopoietic manifestations, and even within the hematopoietic compartment, there is increasing evidence that genes previously thought to be platelet-restricted in their effects may actually have a broader impact on overall blood cell formation. Although many of the inherited thrombocytopenia syndromes are rare, dissecting their genetic underpinning has greatly contributed to our understanding of basic megakaryocyte and platelet biology.
Megakaryopoiesis/thrombopoiesis and the inherited thrombocytopenias
Bone marrow–resident hematopoietic stem cells (HSCs) are multipotent cells with self-renewal capacity able to generate all mature blood lineage cells in a process termed hematopoiesis. Traditional models portray a hierarchy of differentiation beginning with a bifurcation between common lymphoid progenitors and common myeloid progenitors, the latter which ultimately give rise to megakaryocyte-erythroid progenitors and subsequently megakaryocytes (MKs).1-4 Over the last decade, this binary model has been challenged by new findings that suggest that there is a subset of HSCs that express von Willebrand factor (VWF) and have a strong MK bias and limited lymphoid potential. More importantly, the VWF+ HSCs can give rise to VWF− HSCs, whereas the opposite is not true, indicating that these cells are high in the hematopoietic hierarchy.5 As MKs differentiate and mature in the bone marrow, they develop polyploidy, increase the numbers of specialized granules and their cytoplasmic volume, extend cytoplasmic extensions, and develop a complex demarcation membrane system.6 The steps governing commitment of megakaryocyte-erythroid progenitors and MK progenitors toward final stages of MK differentiation are highly regulated. This process initially requires thrombopoietin (TPO) engaging its receptor MPL, leading to downstream JAK2-mediated signaling.7,8 Although most steps in MK maturation involve TPO, it is not essential for final MK maturation and subsequent thrombopoiesis, the process by which proplatelets and ultimately platelets are formed.9 Several transcription factors have been identified as crucial in megakaryopoiesis/thrombopoiesis. Additionally, the formation of proplatelets at the demarcation membrane system occurs only after mature megakaryocytes migrate to and extend protrusions through the vascular sinusoidal space. Under sheer stress from vascular blood flow, platelets are released.6,10 This migration and the dynamic, reversible growth and extension of proplatelet processes requires extensive cytoskeletal reorganization11 and dynamic remodeling, which involves a variety of molecules including CDC42, PAK2, ADF/COFILIN, β1-tubulin, WASP, and many others.12 The average platelet lifespan is ∼7 to 10 days13 with peripheral clearance in part mediated by programmed anuclear cell death and loss of sialic acid with subsequent clearance by Kupffer cells in liver sinusoids.14-16 Therefore, variants in genes involved in every step of these complex processes can cause inherited thrombocytopenia (Figure 1).
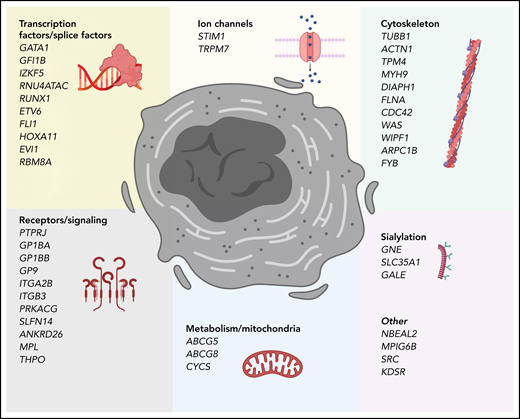
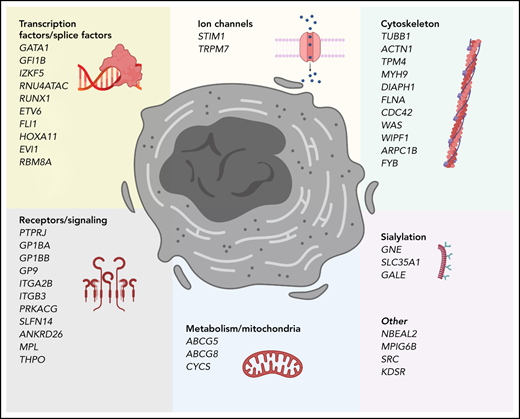
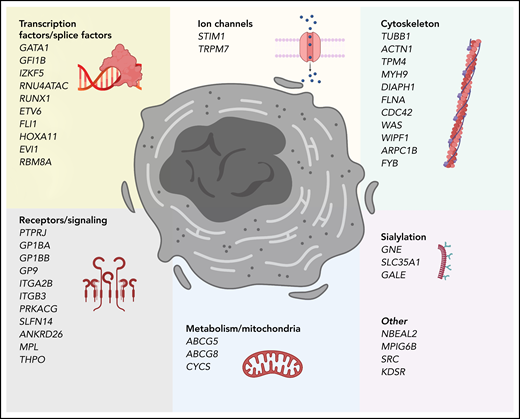
Genes that cause inherited thrombocytopenias grouped by established and potential cellular mechanisms involved in megakaryocyte biology. These genes also correspond to the tier 1 and tier 2 gene lists curated by the International Society of Thrombosis and Hemostasis Genomics in Hemostasis Subcommittee.
Genes that cause inherited thrombocytopenias grouped by established and potential cellular mechanisms involved in megakaryocyte biology. These genes also correspond to the tier 1 and tier 2 gene lists curated by the International Society of Thrombosis and Hemostasis Genomics in Hemostasis Subcommittee.
The inherited thrombocytopenias are an expanding group of disorders17-20 characterized by familial thrombocytopenia and bleeding tendency of various severity with either small, normal, or large sized platelets (Table 1). In some disorders, the defect may be quantitative only, whereas in others, there may be qualitative/functional defects as well. Patients may be identified in the newborn period with easy bruising, petechiae, mucosal bleeding, and thrombocytopenia. However, this constellation of symptoms can also be observed in the much more common allo- and autoimmune thrombocytopenia disorders and in infection- or drug-induced thrombocytopenia. Some of the more common inherited thrombocytopenias are associated with enlarged platelet size (macrothrombocytopenia) or notably small platelet size (microthrombocytopenia); however, as the list of inherited thrombocytopenias expands, so too does the recognition of disorders with platelets that would appear normal in size and morphology on routine peripheral blood smear. Obvious family history of thrombocytopenia should prompt the clinician to consider an underlying genetic etiology, though de novo variants, especially in autosomal dominant syndromes, and the lack of symptomatic relatives as often occurs in recessive disorders means that an absence of family history should not exclude consideration of these entities. Finally, persistent thrombocytopenia, extrahematopoietic abnormalities, bleeding or bruising out of proportion to the degree of thrombocytopenia, refractoriness to medical treatments typically used in immune-mediated thrombocytopenia, and nonresponse to splenectomy should prompt evaluation for an underlying genetic cause.
Diagnostic evaluations
The need to correctly identify patients with inherited thrombocytopenias is pressing. First, many of these patients have extrahematopoietic phenotypes that themselves may require additional medical treatment. Second, identification can help guide proper management. In some individuals, their inherited thrombocytopenia may be mistaken for immune thrombocytopenia, prompting unnecessary splenectomy. Not only will the patient experience persistent thrombocytopenia but would now be at risk for postoperative complications including life-long increased risk for infection. Additional clinical implications are the ability to provide perioperative guidance for necessary surgeries, including the use of antifibrinolytics, and understanding the role and timing of HSC transplant or gene therapy. Although most of the inherited thrombocytopenias do not have a specific treatment, the importance of a precise diagnosis in some of them is critical. For example, the diagnosis of congenital amegakaryocytic thrombocytopenia due to deleterious variants in MPL or THPO will prompt referral for a bone marrow transplant as the only curative option for these patients. Finally, some inherited thrombocytopenia syndromes are associated with an increased risk of hematopoietic malignancy. Although prospective data validating the ability of screening to impact hematopoietic malignancy outcomes in these populations are lacking, a germline diagnosis can help clinicians provide genetic counseling regarding personal risk and family planning.
Diagnostic evaluation of the thrombocytopenic patient with suspected inherited disorder should begin with a thorough history and physical examination. Key history elements include duration of thrombocytopenia, response to previous therapies, other medical history (especially of hearing or vision abnormalities and kidney, heart, or neurologic disease), review of growth curves, and family history including of hematopoietic malignancy. Physical exam should include thorough review of systems involved in specific syndromes including careful examination of the skin and musculoskeletal, cardiac, and neurologic systems (Figure 2). Initial laboratory evaluation starts with a complete blood count using an electronic counter to calculate platelet count and size and to determine red cell indices. However, caution must be used in patients with platelet macrocytosis as cell type can be incorrectly assigned, leading to inaccurate estimate of both platelet number and volume.21 The peripheral smear should be reviewed under routine light microscopy by an experienced hematologist. Special attention should be paid to size, shape, number, and granule appearance. Review should not be limited to the platelet compartment as several inherited thrombocytopenias may demonstrate abnormalities in other blood cell types. For example, in X-linked thrombocytopenia with thalassemia due to variants in GATA1, patients can have microcytosis and reticulocytosis evidenced by polychromasia and anisocytosis.22 Giant platelets, those larger than the size of a normocytic red blood cell, can be observed in Bernard-Soulier syndrome23 and MYH9-related disease, the latter of which can also display leukocyte cytoplasmic inclusions termed Döhle-like body inclusions.24 A paucity of α granules, as can be observed in gray platelet syndrome,25-27GFI1b-related thrombocytopenia,28,29 or SRC-related thrombocytopenia,30,31 can give platelets a “pale” appearance in addition to platelet macrocytosis. In FLI1-associated thrombocytopenia32-34 or thrombocytopenia caused by deletions in 11q23,35,36 some patients’ platelets demonstrate a single, condensed-appearing granule. Small platelets may be observed in Wiskott-Aldrich syndrome, X-linked thrombocytopenia, ARPC1B-related thrombocytopenia, or FYB-related thrombocytopenia. Platelet aggregation can further suggest specific etiologies, for example demonstrating an increased response to ristocetin in platelet-type von Willebrand disease; however, there can be heterogeneity in platelet aggregation findings, and these studies may be most useful to support the suspicion of an inherited thrombocytopenia over a diagnosis of ITP.37 Additional functional assays may have more limited availability given requirements for specialized laboratory testing. This includes platelet glycoprotein expression by flow cytometry, which is mostly used to confirm the diagnosis of Bernard-Soulier syndrome in patients with macrothrombocytopenia and Glanzmann thrombasthenia in patients with absent aggregation and normal platelet count, and whole-mount transmission electron microscopy (TEM) to evaluate dense granule deficiency, as well as thin section TEM for α granule evaluation and other platelet structural abnormalities. Although whole-mount TEM usually confirms the diagnosis of Hermansky-Pudlak syndrome and other dense granule deficiencies,38 thin section TEM provides useful information for the diagnosis of NBEAL2-, GFI1B-, FLI1-, and STIM1-related thrombocytopenias.
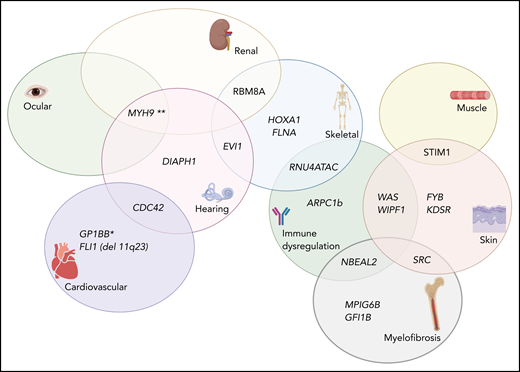
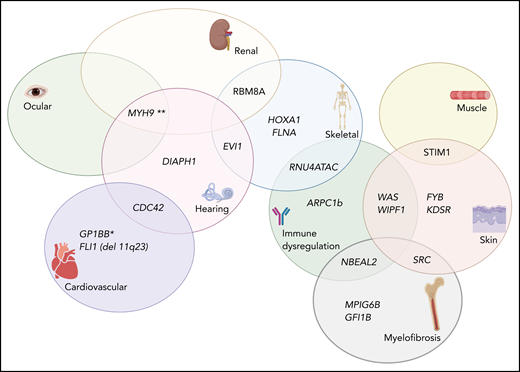
Complex Venn diagram showing the extrahematopoietic manifestations of select germline inherited thrombocytopenia syndromes. *The role of GP1BB in the thrombocytopenia associated with 22q11 del has been recently disputed (see Zwifelhofer et al42).
Complex Venn diagram showing the extrahematopoietic manifestations of select germline inherited thrombocytopenia syndromes. *The role of GP1BB in the thrombocytopenia associated with 22q11 del has been recently disputed (see Zwifelhofer et al42).
Some specific extrahematopoietic findings may also allow the clinician to further narrow the diagnostic possibilities. For example, several inherited thrombocytopenia–associated genes comprise a syndrome that includes radioulnar dysostosis (Table 1). Interestingly, this includes many transcription factors important for megakaryopoiesis including FLI1, HOXA11, MECOM, and RBM8A. A family history of hematopoietic malignancy should prompt consideration of thrombocytopenia related to variants in RUNX1, ETV6, or ANKRD26. Sensorineural hearing loss is a feature of MYH9-, DIAPH1-, and CDC42-associated thrombocytopenia, all of which are implicated in cytoskeletal function. Interestingly, germline variants in several genes have been implicated in promoting increased platelet clearance including FYB, GP1BA-gain-of-function, STIM1, GNE, and WAS. Indeed, splenectomy has been shown to improve platelet counts in patients with Wiskott-Aldrich syndrome presumably by eliminating the main site for platelet clearance.39 Lastly, patients with 22q11 deletion syndrome may experience thrombocytopenia of varying severity together with other canonical features such as facial dysmorphism, hypocalcemia, athymia, congenital heart disease, and recurrent infections.40,41 Although the deletion typically encompasses the critical platelet signaling receptor GP1BB, the role of hemizygosity at this region for thrombocytopenia and bleeding has recently been called into question.42 Furthermore, immune cytopenias responsive to immunomodulation have been previously documented in this patient population,43,44 confounding the ontogeny of their platelet phenotypes.
In a recent excellent review, Pecci and Balduini provide relative frequencies of the genetic etiology behind identifiable inherited thrombocytopenias in a large 335-family collection from Italy.45 First, they note that in their estimation, ∼50% of patients with high suspicion for an inherited thrombocytopenia will not have a known, identifiable underlying cause. Of those families where a more precise genetic diagnosis is made, the combination of features discussed above plus medical history and physical examination could provide high diagnostic support for an inherited thrombocytopenia in over half of patients. However, ultimately, the majority of pathogenic genetic variants may present as isolated thrombocytopenia (Table 1).
Genetic diagnosis, cost, and ethical considerations
Since the application of next-generation sequencing (NGS) to patients with inherited thrombocytopenias, there has been an explosion of newly identified causative genes. Indeed, in the 5-year period from 2015-2019, at least 20 distinct genetic entities causing inherited thrombocytopenia were identified. Several recently identified thrombocytopenia genes may have normal sized platelets, no distinctive morphology on review of peripheral smear, and are not associated with other hematopoietic or extrahematopoietic organ system involvement such as with IZKF5-,46ETV6-,47-50 and THPO-related thrombocytopenia. These examples highlight how NGS can provide diagnostic clarity that may change management, for example advising on and screening for malignancy risk in ETV6, RUNX1, and ANKRD26 patients and their families. Indeed, NGS is now being used in many centers as an up-front component in the evaluation of inherited thrombocytopenias. In the United States, several NGS-targeted panels are available (including Versiti, 23-gene panel; Prevention Genetics, 30-gene panel; Blueprint Genetics, 37-gene panel; and several academic hospital–based clinical laboratories). Of note, when ordering panel-based testing, it is important to ensure that known noncoding pathogenic variants are covered, for example 5′ UTR variants in ANKRD26. Additionally, although NGS can detect small deletions, larger structural variants are missed; therefore, if those are suspected, other techniques such as Multiplex ligation-dependent probe amplification or array comparative genomic hybridization should be used. It is also important to mention that most commercial panels use software-automated reporting, therefore potentially missing small insertions and deletions. As highlighted in Table 1, none of these targeted panels incorporate all known causes of inherited thrombocytopenia, creating an inherent risk for false reassurance in the setting of “negative” genetic testing. Furthermore, it has been shown that the diagnostic yield of these genetic panels is much lower than expected.51 Recently, Downes et al showed that in a large cohort of well phenotyped patients with disorders of bleeding and thrombosis, the diagnostic rate for patients with thrombocytopenia or a known disorder of platelet function was almost 50% and 26%, respectively, findings that were supported in another report by Bastida et al, underscoring the importance of adequate clinical and laboratory phenotyping.52
The genes that populate these panels are selected based on previous discoveries; however, as more patients are sequenced, new variants will be reported for whom it is difficult to assign pathogenicity. Indeed, there is a roughly linear relationship between the number of genes on an NGS-based panel and the number of variants of uncertain significance reported per patient.53 And despite guidelines for variant classification outlined in the critical American College of Medical Genetics/Association for Molecular Pathology standards,54 there are disease contexts for which rarity, benign variation, phenotypic variability, and other potential confounders have generated interlaboratory discordance in variant interpretation. In these contexts, including in the familial platelet disorders, there has been a clear need for expert review of variants. The goal is that systematic curation of these variants can allow for the determination of strong variant-disease associations. Recently, 2 international expert panels, the ClinGen Platelet Gene Curation Expert Panel55 and the International Society on Thrombosis and Haemostasis Subcommittee on Genomics in Thrombosis and Hemostasis,56 reported their initial curation efforts with the ultimate goal of sharing the information publicly and in real time for use by clinicians that work with patients with inherited disorders of hemostasis including inherited thrombocytopenias.
One challenge in the United States is the highly variable access to insurance coverage for NGS-based testing. For example, the Center for Medicare and Medicaid Service supports NGS testing nationally only for the indications of assessing germline breast and ovarian cancer risk.57 Other germline indications are treated as “nationally noncovered.” Current estimates are that 80% of insured individuals have coverage for targeted sequencing panels, which drops to only 56% for Medicaid enrollees.58 Several aspects of genetic testing for inherited thrombocytopenia syndromes raise ethical questions beyond cost. For patients without a family history of hematopoietic malignancy, it is crucial to explain the potential to identify variants in leukemia-associated genes such as RUNX1, ETV6, or ANKRD26. Understanding the purpose of testing for a minor is also critically important: does the testing serve a critical diagnostic function and therefore can be performed after acquiring informed consent, or does the testing serve a predictive purpose that will not change clinical management during the pediatric age range and therefore should be deferred?59 Finally, clinicians should be experienced in the interpretation and counseling of results, including variants of uncertain significant or variants with implications for disease carrier status. For a further review of this topic, please see the recent guideline for ethical considerations of genetic testing in inherited platelet disorders.60
Variant interpretation
Exome sequencing has played a major role in the identification of pathogenic variants in inherited thrombocytopenias. However, as mentioned before, variant interpretation, especially for novel variants, requires careful curation and, if available, access to additional family members to facilitate the correct diagnosis. Additionally, insurance coverage for exome sequencing is estimated at only 63% of insured persons, dropping to 39% for patients with Medicaid. Although research-based exome or whole-genome sequencing (WGS) has led to an explosion in newly identified variants, there are several important considerations that can guide clinical and discovery-based interpretation including phenotyping and functional validation of variants.
Consider the discovery of GATA1 variants and their role in thrombocytopenia syndromes (Table 2). One of 6 members of the GATA transcription factor family, GATA1 contains 2 N-terminal transcriptional activation domains and C-terminal zinc fingers responsible for binding its consensus (A/T)GATA(A/G) sequence (reviewed in Crispino et al61). Although constitutive loss of GATA1 in mice is embryonic lethal,62 an inducible model highlighted its requirement for normal and stress erythropoiesis and demonstrated profound thrombocytopenia.63 Germline variants in GATA1 are linked to a variety of human diseases with aberrant hematopoiesis. An X-linked form of thrombocytopenia with globin chain imbalance resembling β-thalassemia was first described in 1977,22 with the causative pathogenic variant confirmed over 25 years later. This study linked the original pedigree to a pathogenic GATA1 change p.R216Q impacting the ability of GATA1 transcription factor to bind DNA.64 These patients demonstrate bone marrow dyserythropoiesis but only mild anemia. Although a second pedigree with the same variant phenocopied the original mild thrombocytopenia, β-thalassemia–like imbalance in globin synthesis, and dyserythropoiesis,65 this latter study also commented on a severe reduction of α-granules as detected on electron microscopy, a finding confirmed in an additional pedigree whose authors elected to term the syndrome “X-linked gray platelet syndrome.”66 Interestingly, a variant affecting the same residue but with a different substitution, p.R216W, has overlapping clinical features with the additional finding of congenital erythropoietic porphyria characterized by cutaneous photosensitivity, hirsutism, and red urine.67 Similarly, although most variants that impact the ability of GATA1 to bind its cofactor FOG1 are characterized by thrombocytopenia with dyserythropoiesis and transfusion-dependent anemia68-72 including the variant p.D218Y,73 overlapping variants p.D218N and p.D218G patients have thrombocytopenia alone without erythrocyte abnormalities.74,75 Truncating GATA1 variants caused by splice site alterations lead to exon 2 skipping and production of an expressed protein that lacks the N-terminal transactivating domain. These patients present with dyserythropoietic, steroid-responsive anemia with clinical features overlapping Diamond-Blackfan anemia (DBA), though only 2 of 5 pedigrees demonstrate elevated erythrocyte adenosine deaminase.76-80 Interestingly, although the various exon 2 bordering splice variants were shown to produce the same short form of GATA1, only 1 pedigree was affected by thrombocytopenia,80 whereas 2 separate families with the identical variant did not.77,78 Across several families, confirmed heterozygous GATA1 females were either asymptomatic or demonstrated mild thrombocytopenia and imbalanced globin chain synthesis reflecting skewed X-inactivation.
This example highlights the challenges that phenotypic variability can pose to the clinician and researcher. However, understanding the phenotypic overlap with DBA has led to further mechanistic insight into DBA pathogenesis. In an elegant study from the Sankaran laboratory, primary hematopoietic cells from patients with DBA and variants in the classic DBA gene RPS19 were shown through polysome profiling to have a specific decrease in GATA1 messenger RNA translation and a decreased amplitude in the GATA1 target gene transcriptional signature.81 Ineffective erythropoiesis in cultured primary cells could be partially rescued by GATA1 overexpression, suggesting potential therapeutic implications.
Platelet phenotype variability as well as incomplete penetrance can also confound novel variant identification. Whole-exome or genome-based NGS strategies to identify novel, rare pathogenic variants can derive increased power by incorporating multiple family members, both affected and unaffected. Especially in families with an apparent autosomal dominant transmission pattern, filtering out variants that are absent from unaffected individuals can be an effective step to narrow variants of interest. Adding to this complexity, in some GATA1 pedigrees, affected individuals can have improvement in their thrombocytopenia with age. Thrombocytopenia can be mild and potentially unrecognized in some family members, for example as seen across several large pedigrees of RUNX1-mutated familial platelet disorder. In the recently described syndrome of IKZF5-related thrombocytopenia,46 rare missense variants from WGS of 105 thrombocytopenic patients were identified through a Bayesian inference framework using over 10 000 unaffected individuals. However, in one pedigree with 6 family members that ultimately underwent analysis for the variant in question, 1 family member carried the proposed pathogenic p.G134E yet had a normal platelet count of 184. The variant otherwise segregated with disease, which was characterized by a mild thrombocytopenia with normal platelet size and mild bleeding symptoms. This is one example of how in whole-exome sequencing/WGS studies of large pedigrees, mildly affected individuals may be phenotyped as “unaffected” and the variant filtered out as not segregating with disease. This emphasizes the need for complete phenotyping on individuals both affected and unaffected, with attention to not only absolute platelet number but also morphology and, when available, functional testing.
Validation of variants
Validation of potential novel variants should be accompanied by functional testing when possible. For decades, investigators have used molecular biology, cell biology and imaging and biochemistry techniques to successfully validate specific genetic variants in megakaryocytes, platelets, and other cell models, including western blots for protein detection, confocal microscopy for cellular defects associated with cytoskeleton genes, and reporter assays for transcription factors. Additionally, animal models of inherited thrombocytopenias have been generated over the years with mixed results. For example, whereas mouse knock-out models for Myh9, Nbeal2, and Gp1bb appear to replicate the phenotype observed in humans, others such as Runx1, Was, and Mpl only replicate certain features of the human disease82,83 but not all. More recently zebrafish models have been successfully used to validate variants in PTPRJ84 and SRC31 and to establish a model of congenital amegakaryocytic thrombocytopenia (MPL).85 Optimally, primary bone marrow hematopoietic tissue from affected patients and normal controls could be compared with assess markers of megakaryocyte maturation and morphology, granule production, and platelet function using platelet aggregation studies, flow cytometry, and other functional methods. However, it is also increasingly recognized that in vitro culture of megakaryocytes has limitations, although some new technologies have allowed investigators to culture megakaryocytes and study their function from minimal volumes of bone marrow aspirates.86
Recently, investigators were able to use induced pluripotent stem cells from patients with ETV6 and RUNX1 variants, as well as introducing the same gene variants by clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 in an isogenic induced pluripotent stem cell line and studied their effect on hematopoietic progenitor and megakaryocyte maturation.87 In the absence of primary patient samples, current technology also allows for the in vitro manipulation of hematopoietic stem and progenitor cells either through overexpression of variant alleles or through genetic deletion via CRISPR/CRISPR-associated protein 9. These cells can be differentiated with TPO along the megakaryocytic pathway to form proplatelet-forming megakaryocyte-like cells, allowing investigators to study the effect of these genetic defects on megakaryocyte differentiation and maturation.88
Structural modeling can be useful in predicting the impacts of variants on protein function; however, limitations exist, including the availability of a 3-dimension model and translation to functional impact. For example, a recent study looking at the impact of a large number of ETV6 variants identified through sequencing of an acute lymphoblastic leukemia cohort developed a reporter system of transcriptional repressor activity.89 Variants predicted to be pathogenic and those that emerged from studies of individual thrombocytopenic/familial leukemia pedigrees do not strictly occur in the region at the ETV6-DNA interface. Emerging technologies such as base editor screens will allow for more rapid variant screening in relevant cellular models.90 It is important to mention that although exome and short-read genome sequencing may allow for discovery of novel missense or small insertion/deletion variants, other genomic technologies are required to detect larger copy number or structural alterations. For example, long-read genomic sequencing facilitated the recent discovery of a structural variant causing a paired-duplication inversion leading to pathogenic gain-of-function WAC-ANKRD26 fusion.91 These variants will not be discernable by currently available clinical testing but may also be overlooked by most research-based sequencing efforts. As costs for long-read genome sequencing and additional advanced genomic techniques come down, the availability of these newer platforms will likely facilitate additional complex structural genomic abnormalities associated with inherited thrombocytopenia syndromes.
Lastly, there is an increasing appreciation for the effects of genes initially identified as being most important for megakaryopoiesis or thrombopoiesis on nonplatelet hematopoietic cells. A clear example is TPO, originally described as a cytokine that stimulates megakaryopoiesis92,93 but later identified as playing an important role in HSC maintenance,94 especially in promoting HSC quiescence.95,96 Patients with Roifman syndrome due to biallelic germline variants in RNU4ATAC also display abnormal differentiation of B cells with associated hypogammaglobulinemia and recurrent viral infections.97 Beyond factors implicated in differentiation, factors associated with platelet differentiation and granule formation such as NBEAL2 have also been shown to impact mast cell differentiation and granule generation98 as well as demonstrating clinical phenotype consistent with broader immune dysregulation.99 This immune dysregulation also occurs in patients with variants in actin cytoskeletal organization genes (Table 1), for example patients with germline variants in ARPC1B who demonstrate megakaryocyte differentiation defects100 but also disruption of T-cell lineage development.101 As additional genes are added to the growing list of thrombocytopenia disorders, careful phenotyping will allow researchers to extend this new knowledge to other hematopoietic compartments.
Conclusions
Over the last 2 decades, the advancement in genomic techniques and decreased cost of sequencing have allowed for the elucidation of the genetic basis of many inherited thrombocytopenias. For most of these newly discovered genes, this was the result of international collaborative efforts that included clinicians, scientists, and statistical geneticists, underscoring the importance of collaboration and team science. Although the sequencing of patients and families that led to these discoveries paved the way for better understanding of megakaryocyte and platelet biology, challenges remain on the validation of genetic variants in available models. Ultimately, this “bedside to bench” approach that moved the field forward will hopefully make its way back to better inform patients about their disease.
Acknowledgments
Figures were created with biorender.com.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants R01 HL120728 and R01 HL141794 (J.D.P.).
Authorship
Contribution: J.T.W. and J.D.P. wrote the manuscript.
Conflict-of-interest disclosure: J.D.P. reports consulting for CSL Behring. J.T.W. declares no competing financial interests.
Correspondence: Jorge Di Paola, Division of Hematology-Oncology, Department of Pediatrics, Washington University School of Medicine, 660 S Euclid Avenue, Campus Box 8208, 5th floor MPRB, St. Louis, MO 63110; e-mail: dipaolaj@wustl.edu.