

In this issue of Blood, Alvarez et al1 present evidence that the epigenetic regulator plant homeodomain finger 6 (PHF6) plays an important role in the prevention of genomic instability in leukemia cells by participating in a wide range of cellular processes that mediate DNA repair and resolution of replication stress.
To protect their genetic information, mammalian cells have evolved a complex network of high-fidelity biological processes to regulate chromatin structures, recognize genomic lesions, and repair damaged DNA. Chromatin regulation involves epigenetic mechanisms such as DNA methylation, histone modifications, and microRNAs, whereas DNA damage response (DDR) relies on a complex network of sensors, transducers, and effectors that coordinate DNA repair with DNA replication. It has recently become clear that epigenetic control and DNA repair cooperate closely. Epigenetic regulators can directly control the expression level and activity of DNA repair proteins, as well as the accessibility of chromatin to the recruited DNA damage repair complexes. It is not surprising, therefore, that important mediators of both DNA repair and epigenetic regulation are frequently altered in cancer cells.2,3
PHF6 is an epigenetic regulator frequently inactivated in T-cell acute lymphoblastic leukemia (T-ALL), mixed-phenotype acute leukemia with T-lineage differentiation, and less frequently in acute myeloid leukemia and other myeloid malignancies. Earlier studies have identified PHF6 as a tumor suppressor and have suggested a role for PHF6 in regulating DNA transcription, DNA repair, and hematopoiesis. However, the precise mechanism underpinning its tumor suppressor role remains unclear.4
In this important work, Alvarez et al unraveled different PHF6 protein interactions and provided compelling evidence that PHF6 plays a much wider role in suppressing tumorigenesis than previously understood. The authors observed that PHF6 interacts not only with components of the nucleosome remodeling deacetylase (NuRD) complex that associates with condensed chromatin but also with switch/sucrose nonfermentable (SWI/SNF) chromatin remodelers that bind to open chromatin, suggesting a broad role of PHF6 in chromatin remodeling. Moreover, PHF6 was demonstrated to interact with a range of DNA repair factors at sites of damaged DNA, as well as with regulators of cell cycle and DNA synthesis. Through elegant functional studies, the authors confirmed the ability of PHF6 to facilitate resolution of both single-strand and double-strand DNA breaks. They also highlighted a novel function of PHF6 in homologous recombination (HR) repair that involved promoting the nuclear retention of the HR protein BRCA1.
The authors further observed that the role of PHF6 includes the epigenetic regulation of DNA replication, whereby PHF6 protects replication fork integrity by facilitating ataxia telangiectasia and Rad3-related protein (ATR)–dependent activation of the single-strand DNA-binding replication protein A (RPA). Other epigenetic regulators are known to regulate DNA repair,2 but remarkably, PHF6 is capable of localizing to difficult-to-replicate DNA regions, such as heterochromatin and heterochromatin-associated common fragile sites (CFSs) that are exceptionally susceptible to replication stress and are well-established hotspots for chromosome breakage and rearrangements.5 Therefore, the colocalization of PHF6 with CFSs underscores its fundamental role in the prevention of genomic instability, in which it synchronizes replication dynamics with DNA repair. Taken together, the findings of Alvarez et al suggest a place for PHF6 at the top of the genome regulatory hierarchy as a master epigenetic regulator of DNA repair (see figure).
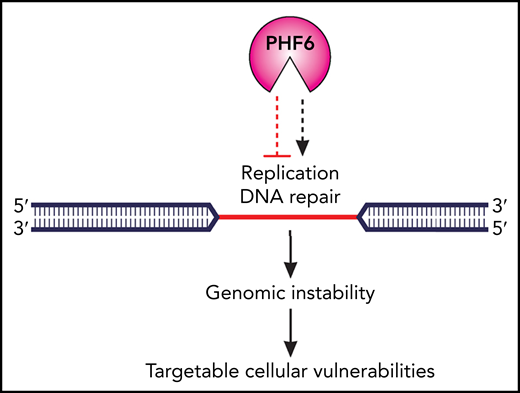
Cellular consequences of the loss of PHF6 function in leukemia cells. Functional loss of the epigenetic regulator PHF6 affects the balance between DNA replication and DNA repair in difficult-to-replicate DNA heterochromatin regions, with subsequent destabilization of common fragile sites and genomic instability. To survive, PHF6-deficient tumor cells must develop adaptive mechanisms and this provides a platform for therapeutic targeting. Professional illustration by Patrick Lane, ScEYEnce Studios.
Cellular consequences of the loss of PHF6 function in leukemia cells. Functional loss of the epigenetic regulator PHF6 affects the balance between DNA replication and DNA repair in difficult-to-replicate DNA heterochromatin regions, with subsequent destabilization of common fragile sites and genomic instability. To survive, PHF6-deficient tumor cells must develop adaptive mechanisms and this provides a platform for therapeutic targeting. Professional illustration by Patrick Lane, ScEYEnce Studios.
What are the translational implications of the discoveries by Alvarez et al? In the era of precision medicine, one approach would entail the use of epigenetic therapies that aim to reverse the aberrant epigenetic programs of PHF6-deficient tumor cells.6 In addition, the concept of synthetic lethality, involving the pharmacologic inhibition of biological pathways that cancer cells are dependent upon for survival, has recently emerged as an important paradigm in cancer treatment.7 Such an approach has been studied across a spectrum of DDR-defective tumors, including hematologic malignancies harboring ataxia-telangiesctasia mutated (ATM) defects.8-10 Indeed, similarity exists between ATM and PHF6 in that both regulate a complex network of cellular pathways and participate in the resolution of DNA damage and replication stress. Consequently, their functional loss in cancer cells is likely to create adaptive mechanisms that may give rise to actionable cellular vulnerabilities.
Recent advances, including CRISPR-based gene editing, have made possible systematic screens for synthetic lethal drug targets in human cancers. Such studies will therefore likely reveal novel therapeutic targets in PHF6-deficient cells. With this in mind, it is important to acknowledge that targetable vulnerabilities might depend on the type of malignancy in which PHF6 function is lost. For example, PHF6 inactivation is an early event in T-ALL leukemogenesis and is associated with NOTCH1mutations or overexpression of TLX1 and TLX3, whereas in myeloid malignancies, PHF6 mutations tend to occur later and are frequently accompanied by RUNX1 mutations.4 Therefore, the cell-specific and genotype-specific contexts are likely to influence tumor adaptive mechanisms, and this should be considered in future studies of PHF6-associated tumor vulnerabilities (see figure).
In conclusion, there is an exciting time ahead for scientists who work on the mechanistic aspects of the interplay between epigenetics and DNA repair and for physicians who will be in a position to develop new treatments for hematologic cancers that harbor epigenetic and DDR defects.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

