In this issue of Blood, Li et al harness an elegant series of preclinical models to address 3 questions in the biology of myeloproliferative neoplasms (MPNs). They ask how the TP53 allelic state promotes leukemic transformation; what the cellular origin of the resultant leukemia is; and whether the TP53 mutant clones bear specific vulnerabilities amenable to therapeutic exploitation.1
Around 10% to 20% of patients with MPNs undergo transformation to acute myeloid leukemia (AML), but with a pathological and clinical phenotype that is strikingly different from de novo AML. Although mutations affecting signaling pathways, such as KRAS, KIT, and FLT3, are late events in de novo AML, post-MPN AML generally arises after long-standing dysregulation of JAK/STAT signaling. Post-MPN AML is also associated with a different spectrum of mutations and has an increased incidence of erythromegakaryoblastic leukemias. These contrasts most likely explain why post-MPN AML responds poorly to traditional AML therapies, resulting in a bleak median survival of ∼6 months with no improvement in outcomes over the past 15 years.2 Allogeneic stem cell transplantation remains the only therapy that can potentially convey a survival benefit, although even with transplantation, few patients live >3 years. Therefore, there is a major need for innovative therapies in this setting.3
Mutations in TP53 are one of the most frequent genetic alternations in all human cancers, are a poor prognostic marker in myeloid malignancies, and are strongly associated with complex karyotypic changes and resistance to conventional therapy. In post-MPN AML, studies have confirmed that (1) TP53 mutations are the most frequent mutations at transformation, occurring in ∼30% of patients4; (2) TP53 mutant clones expand at the time of transformation, frequently in association with copy number alterations (CNAs) in 17p and concomitant loss of the wild-type (WT) allele5; and (3) deletion of Trp53, combined with a Jak2V617F (VF) mutation in murine models, leads to a highly penetrant myeloid leukemia.6,7 A study of >3000 patients with myelodysplastic syndromes emphasized the importance of the TP53 allelic state on outcome, demonstrating that high-risk disease was restricted to patients with biallelic TP53 loss, whereas patients with monoallelic aberrations had a clinical course similar that of those with WT TP53.8
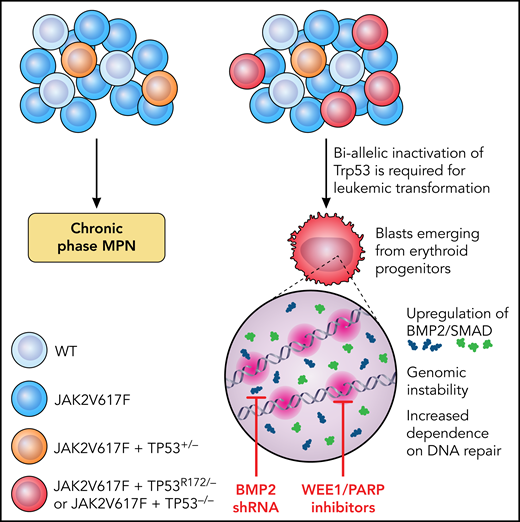
Why TP53 has such a significant impact on disease progression, and why the allelic state in MPNs is so important are incompletely understood. Li et al set out to investigate the impact of TP53 mutations and allelic configuration on leukemic transformation (LT) in MPN and to identify potentially translatable targets (see figure). They generated a conditional oncogenic allele of Trp53 (PR172H) and a conditional loss-of-function allele of Trp53 (P−/−) and created an allelic series of single-mutant (VF) and double-mutant (VF-P) mice. Mice with homozygous p53 loss on a background of VF rapidly transformed to leukemia with a “pure erythroid” subtype (PEL) and were moribund within 8 to 13 weeks. In contrast, VF mice that retained 1 WT copy of Trp53 had a longer survival without LT, confirming the essentiality of biallelic TP53 inactivation for progression.
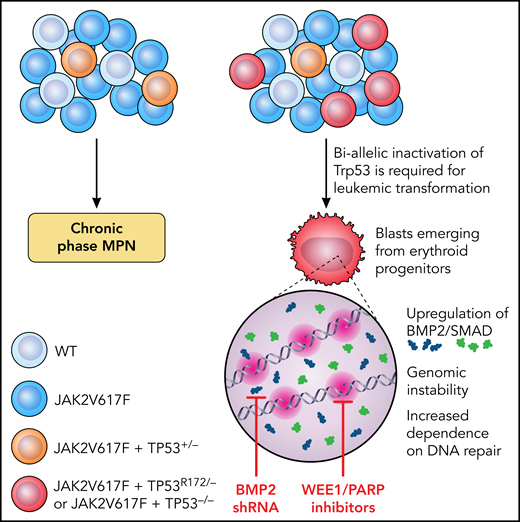
Schematic showing the requirement for biallelic inactivation of Trp53 for leukemic transformation in JAK2V617F-mutant MPNs. Erythroid progenitors appear uniquely vulnerable to Trp53 loss, resulting in upregulation of BMP2/SMAD signaling and aberrant self-renewal and leukemogenic potential. The genomic instability resulting from JAK2V617F plus TP53−/− caused an increased dependence on DNA repair mechanisms and a therapeutic vulnerability to WEE1 and PARP inhibitors. Illustration by Patrick Lane, ScEYEnce Studios.
Schematic showing the requirement for biallelic inactivation of Trp53 for leukemic transformation in JAK2V617F-mutant MPNs. Erythroid progenitors appear uniquely vulnerable to Trp53 loss, resulting in upregulation of BMP2/SMAD signaling and aberrant self-renewal and leukemogenic potential. The genomic instability resulting from JAK2V617F plus TP53−/− caused an increased dependence on DNA repair mechanisms and a therapeutic vulnerability to WEE1 and PARP inhibitors. Illustration by Patrick Lane, ScEYEnce Studios.
Li et al also identified the hematopoietic stem and progenitor cell (HSPC) compartment most vulnerable to TP53-induced LT. They found gross expansion of immunophenotypic megakaryocyte-erythroid progenitors (MEPs) at the time of LT and showed that the expanded population corresponded to erythroid-primed progenitors by single-cell RNA sequencing, with aberrant enrichment of stem-cell–like features coupled with leukemia-initiating capacity after transplant. P-R172H/P- and P−/− blasts displayed common transcriptional alterations, with prominent upregulation of TGF-β/BMP pathways, a finding validated in primary samples from patients with PEL and post-MPN AML. A functional role in post-MPN leukemogenesis was confirmed as short-hairpin RNA knockdown of BMP2 (but not BMP4) impaired leukemic MEP self-renewal and leukemic potential, thereby enhancing murine survival.
JAK2V617F results in a replication stress that is exacerbated by TP53 mutations. Li et al observed that blasts with biallelic TP53 loss incurred frequent CNAs and activation of DNA damage-response pathways. They postulated that this effect causes therapeutic vulnerability related to selective dependency on the remaining DNA-repair pathways pCDC2 and PARP to guard genomic integrity. Treatment with a synergistic combination of the WEE1 and PARP inhibitors adavosertib and olaparib promoted DNA damage and apoptosis, reducing leukemic burden and prolonging survival in mice compared with vehicle or single-agent–treated controls.
Li et al thus generated preclinical models that faithfully recapitulate key aspects of post-MPN AML, in a field where preclinical models and effective therapies have been sorely lacking. Their observations advance our understanding of TP53 biology, confirming the need for biallelic loss and showing that in the context of mutant Jak2, Trp53 mutant, and null genotypes share molecular signatures and clinical course. A key novel finding of this study is that erythroid progenitors are uniquely vulnerable to TP53-mediated leukemogenesis. They provided tantalizing preclinical evidence of efficacy of the dual inhibition of PARP and WEE1. Further validation in patient samples is necessary, but the identification of a novel therapeutic approach with orally bioavailable compounds known to be tolerated from solid organ cancer trials9,10 is certainly compelling.
The findings of Li et al inspire numerous questions for future studies. Why does a JAK2 mutant background promote emergence of TP53 mutations and compound their impact? How do complex cytogenetic changes contribute to LT? The observation that post-MPN AML emerges from the erythroid compartment is intriguing. Is the erythroid stage uniquely vulnerable to the TP53 KO effects, or does TP53-KO induce an erythroid bias and differentiation block? Mechanistic links between BMP2/SMAD and DNA damage and the impact of BMP2/SMAD inhibition await further confirmation. Along with BMP2/SMAD, key inflammatory pathways such as NF-κB and tumor necrosis factor-α were also upregulated in Trp53 KO/mutant blasts. The interplay between cell-extrinsic processes, such as inflammation with clonal evolution, is fascinating, and has been understudied to date. Finally, TP53-mediated LT is but one route of leukemogenesis in MPN. The >60% of post-MPN AML cases that are not TP53 mutant, but often are enriched for epigenetic modifiers (eg, TET2, ASXL1, IDH1/2, and EZH2), raise questions about how DNA methylation and chromatin dynamics play out in MPN clonal expansion. This is another mystery, and one where advances in preclinical models could also be hugely informative.
Conflict-of-interest disclosure: B.P. has had paid speaking engagements for Novartis; consulted for Constellation Therapeutics; received research funding from Galecto Biotech; and is a cofounder and consultant for Alethiomics, holding share options and receiving research funding. C.B. declares no competing financial interests.