


Abstract
Complement is an elaborate system of innate immunity. Genetic variants and autoantibodies leading to excessive complement activation are implicated in a variety of human diseases. Among them, the hematologic disease paroxysmal nocturnal hemoglobinuria (PNH) remains the prototypic model of complement activation and inhibition. Eculizumab, the first-in-class complement inhibitor, was approved for PNH in 2007. Addressing some of the unmet needs, a long-acting C5 inhibitor, ravulizumab, and a C3 inhibitor, pegcetacoplan, have also now been approved for PNH. Novel agents, such as factor B and factor D inhibitors, are under study, with very promising results. In this era of several approved targeted complement therapeutics, selection of the proper drug must be based on a personalized approach. Beyond PNH, complement inhibition has also shown efficacy and safety in cold agglutinin disease, primarily with the C1s inhibitor of the classical complement pathway sutimlimab, as well as with pegcetacoplan. Furthermore, C5 inhibition with eculizumab and ravulizumab, as well as inhibition of the lectin pathway with narsoplimab, is being investigated in transplantation-associated thrombotic microangiopathy. With this revolution of next-generation complement therapeutics, additional hematologic entities, such as delayed hemolytic transfusion reaction or immune thrombocytopenia, might also benefit from complement inhibitors. Therefore, this review aims to describe state-of-the-art knowledge of targeting complement in hematologic diseases, focusing on (1) complement biology for the clinician, (2) complement activation and therapeutic inhibition in prototypic complement-mediated hematologic diseases, (3) hematologic entities under investigation for complement inhibition, and (4) other complement-related disorders of potential interest to hematologists.
Introduction
Complement is an elaborate system involved in innate immune response. Genetic variants and autoantibodies leading to unregulated complement activation are implicated in the pathogenesis of various human diseases. Among them, paroxysmal nocturnal hemoglobinuria (PNH) remains the prototypic model of complement activation and inhibition.1 Eculizumab, the first-in-class complement inhibitor, was first approved for PNH and then approved for complement-mediated diseases/complementopathies across specialties, including atypical hemolytic uremic syndrome (HUS), myasthenia gravis, and neuromyelitis optica spectrum disorder.2 Establishing the clinical merit of targeting complement, preclinical and clinical studies have not only revealed the unmet needs paving the way for next-generation complement therapeutics but also identified other diseases that might benefit from complement inhibition.3
In light of recent advances, this review aims to describe state-of-the-art knowledge of targeting complement in hematologic diseases, focusing on (1) complement biology for the clinician, (2) complement activation and inhibition in prototypic complement-mediated hematologic diseases, (3) hematologic entities under investigation for complement inhibition, and (4) other complement-related disorders of potential interest to hematologists.
Complement biology for the clinician
Complement is composed of >50 proteins providing innate defense against microbes and mediating inflammatory responses.4,5 Because complement is in a constant low-level activation triggered by spontaneous C3 hydrolysis, excessive complement activation is physiologically prevented by membrane-bound or soluble complement regulatory proteins that play an important role in the pathogenesis of complement-mediated diseases. Figure 1 summarizes the traditionally described pathways of complement activation, including the classical, alternative, and lectin pathways. Additional direct interactions between complement and coagulation are mediated by various complement proteins and other coagulation factors, which in turn activate complement.6-11 Thrombin also acts as a C5a convertase,12 while complement and platelets interact during the early atherogenetic process.13,14 Interestingly, indirect effects of complement on thrombosis have been also observed in hemolytic anemias.15 In line with clinical evidence presented in the following section, recent data suggest that C5 inhibition significantly attenuates heme-induced thromboinflammation.16
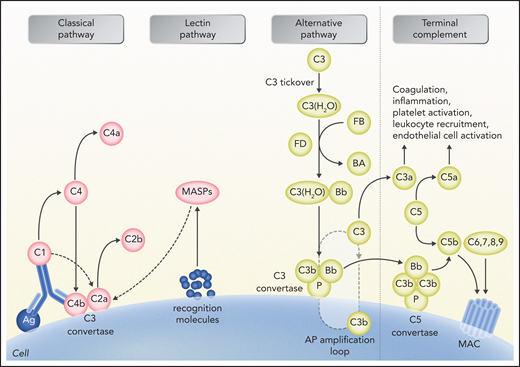
Schematic representation of complement activation and interactions with other systems. The alternative pathway (AP) is constantly activated through slow spontaneous hydrolysis of C3 forming C3(H2O) (ie, tickover). Activated C3(H2O) pairs with factor B, generating C3(H2O)B. Factor B is then cleaved by factor D and generates the fluid phase AP C3 convertase, or C3(H2O)Bb. The latter catalyzes the cleavage of additional C3 molecules to generate C3a and C3b. C3b binds factor B on cell surfaces, which is subsequently cleaved by factor D to generate a second (surface phase) AP C3 convertase (C3bBb), initiating the amplification loop. Binding and cleavage of an additional C3 to C3 convertase form the APC C5 convertase (C3bBbC3b) that cleaves C5 to C5a and C5b. Both C3 and C5 AP convertases are stabilized by properdin, or factor P, which also serves as a selective pattern recognition molecule for de novo C3 AP convertase assembly. Classical pathway activation mainly depends on antibody-antigen complexes recognized via complement component C1q. C1q also binds directly to certain epitopes from microorganisms or apoptotic cells and to cell-surface molecules. C1q then cleaves C1r, which activates C1s protease. Subsequently, C1s cleaves C4 and C2, leading to the formation of classical pathway C3 convertase (C4bC2a). C3 convertase cleaves C3, generating the anaphylatoxin C5a and C5 convertase (C4bC2aC3b), which cleaves C5 into C5a and C5b, which initiate the terminal pathway of complement. Lectin pathway activation is initiated by recognition of carbohydrate structures on the microbial surfaces by mannose-binding lectins (MBLs). Additional pattern recognition molecules of the lectin pathway include ficolins and collectin 11. These molecules act through MBL-associated serine proteases (MASPs), generating C3 convertase (C4bC2a), similarly to the activation of classical pathway. Proximal complement activation initiated by any of the 3 pathways (classical, alternative, or lectin) leads to C3 activation and C3 convertase formation on C3-opsonized surfaces. In the presence of increased surface density of deposited C3b, C5b initiates the terminal complement pathway, binding to C6 and generating C5b-6, which in turn binds to C7, creating C5b-7. C5b-7 is able to insert itself into lipid layers of the membrane. Once there, C5b-7 binds C8 and C9, forming a complex that unfolds in the membrane and binds several C9 molecules, thereby forming the membrane attack complex (MAC) on the surface of target cells. C3a and C5a mediate complement interactions with inflammation, coagulation, platelet activation, leukocyte recruitment, and endothelial cell activation. Professional illustration by Somersault 18:24.
Schematic representation of complement activation and interactions with other systems. The alternative pathway (AP) is constantly activated through slow spontaneous hydrolysis of C3 forming C3(H2O) (ie, tickover). Activated C3(H2O) pairs with factor B, generating C3(H2O)B. Factor B is then cleaved by factor D and generates the fluid phase AP C3 convertase, or C3(H2O)Bb. The latter catalyzes the cleavage of additional C3 molecules to generate C3a and C3b. C3b binds factor B on cell surfaces, which is subsequently cleaved by factor D to generate a second (surface phase) AP C3 convertase (C3bBb), initiating the amplification loop. Binding and cleavage of an additional C3 to C3 convertase form the APC C5 convertase (C3bBbC3b) that cleaves C5 to C5a and C5b. Both C3 and C5 AP convertases are stabilized by properdin, or factor P, which also serves as a selective pattern recognition molecule for de novo C3 AP convertase assembly. Classical pathway activation mainly depends on antibody-antigen complexes recognized via complement component C1q. C1q also binds directly to certain epitopes from microorganisms or apoptotic cells and to cell-surface molecules. C1q then cleaves C1r, which activates C1s protease. Subsequently, C1s cleaves C4 and C2, leading to the formation of classical pathway C3 convertase (C4bC2a). C3 convertase cleaves C3, generating the anaphylatoxin C5a and C5 convertase (C4bC2aC3b), which cleaves C5 into C5a and C5b, which initiate the terminal pathway of complement. Lectin pathway activation is initiated by recognition of carbohydrate structures on the microbial surfaces by mannose-binding lectins (MBLs). Additional pattern recognition molecules of the lectin pathway include ficolins and collectin 11. These molecules act through MBL-associated serine proteases (MASPs), generating C3 convertase (C4bC2a), similarly to the activation of classical pathway. Proximal complement activation initiated by any of the 3 pathways (classical, alternative, or lectin) leads to C3 activation and C3 convertase formation on C3-opsonized surfaces. In the presence of increased surface density of deposited C3b, C5b initiates the terminal complement pathway, binding to C6 and generating C5b-6, which in turn binds to C7, creating C5b-7. C5b-7 is able to insert itself into lipid layers of the membrane. Once there, C5b-7 binds C8 and C9, forming a complex that unfolds in the membrane and binds several C9 molecules, thereby forming the membrane attack complex (MAC) on the surface of target cells. C3a and C5a mediate complement interactions with inflammation, coagulation, platelet activation, leukocyte recruitment, and endothelial cell activation. Professional illustration by Somersault 18:24.
Complement activation in hematologic diseases
PNH
PNH is an acquired hematologic disease resulting from loss-of-function variants in the PIGA gene17 occurring in ≥1 hematopoietic stem cells, which eventually expand over normal hematopoiesis.18 All progeny blood cells carry the aberrant phenotype characterized by lack of surface glycosylphosphatidylinositol-linked proteins, including 2 endogenous complement regulators, CD5919 and CD55.20 As a consequence, PNH erythrocytes are unable to modulate complement activation on their surface, and the continuous, spontaneous C3 tickover21 occurring in the fluid phase leads to surface complement activation, with generation of glycophorin-bound alternative pathway C3 convertase.22 Once C5 convertases are generated as described in “Complement biology for the clinician” (Figure 1), the terminal complement pathway is initiated, which eventually leads to MAC assembly resulting from lack of CD59.19 These events account for MAC-mediated intravascular hemolysis, which is the hallmark of PNH.23 As discussed in “Complement inhibition in hematologic diseases,” the terminal complement pathway has become a therapeutic target24 for PNH treatment, with remarkable clinical results.25-28 On the other hand, clinical use of terminal complement has drawn attention to the fact that complement dysregulation in PNH is broader and not limited to the terminal pathway. Indeed, we have demonstrated that, during anti-C5 therapy, early complement activation remains uncontrolled on PNH erythrocytes, leading to surface C3 activation and progressive opsonization, with C3 fragments of PNH erythrocytes spared from MAC-mediated hemolysis.29,30 Because C3d-opsonized erythrocytes may be recognized by professional macrophages through C3dg receptors,31 C3-mediated extravascular hemolysis has emerged as an additional/alternative mechanism of hemolysis in patients with PNH receiving anti-C5 therapy.29,30,32,33Figure 2 summarizes the mechanisms for C5 inhibition failure. Thus, different mechanisms of complement-mediated damage may contribute to disease manifestations in PNH, possibly indicating a scenario that is more complex than anticipated.
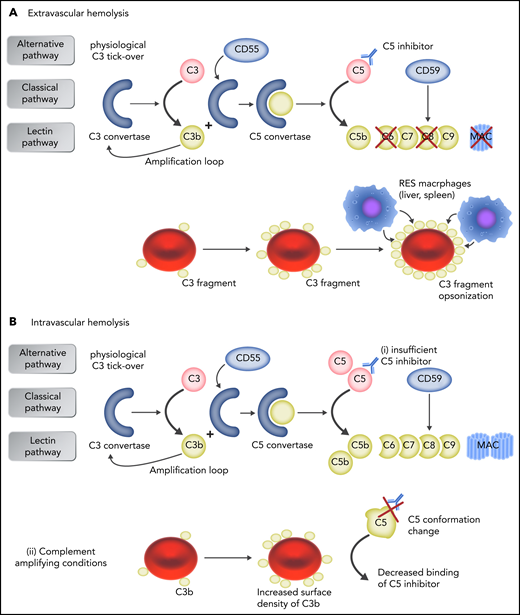
Mechanisms of failure to C5 inhibition. (A) In patients who receive anti-C5 therapy, early complement activation remains uncontrolled on PNH erythrocytes, leading to surface C3 activation and progressive opsonization, with C3 fragments of PNH erythrocytes spared from MAC-mediated hemolysis. Because C3d-opsonized erythrocytes may be recognized by professional macrophages through C3dg receptors, C3-mediated extravascular hemolysis has emerged as additional/alternative mechanism of hemolysis in patients with PNH receiving anti-C5 therapy. (B) Intravascular hemolysis is caused either by insufficient drug dosing, allowing free C5 levels to rise (i), or by complement-amplifying conditions (eg, pregnancy, infection, major surgery), resulting in excess C3b accumulation on PNH erythrocytes, which decreases the binding of C5 inhibitor to C5 because of conformation change in C5 and generates high-affinity, C3b-rich, C5 convertases that compete more efficiently with anti-C5 antibodies for their substrate C5 (ii). Professional illustration by Somersault 18:24.
Mechanisms of failure to C5 inhibition. (A) In patients who receive anti-C5 therapy, early complement activation remains uncontrolled on PNH erythrocytes, leading to surface C3 activation and progressive opsonization, with C3 fragments of PNH erythrocytes spared from MAC-mediated hemolysis. Because C3d-opsonized erythrocytes may be recognized by professional macrophages through C3dg receptors, C3-mediated extravascular hemolysis has emerged as additional/alternative mechanism of hemolysis in patients with PNH receiving anti-C5 therapy. (B) Intravascular hemolysis is caused either by insufficient drug dosing, allowing free C5 levels to rise (i), or by complement-amplifying conditions (eg, pregnancy, infection, major surgery), resulting in excess C3b accumulation on PNH erythrocytes, which decreases the binding of C5 inhibitor to C5 because of conformation change in C5 and generates high-affinity, C3b-rich, C5 convertases that compete more efficiently with anti-C5 antibodies for their substrate C5 (ii). Professional illustration by Somersault 18:24.
Cold agglutinin disease
Cold agglutinin disease (CAD) is a clonal B-cell lymphoproliferative disorder, whereas secondary CAD is associated with infectious and neoplastic disorders, such as lymphomas and carcinomas.34 Patients with CAD have a high risk of thromboembolism and early mortality. The disease is typically caused by immunoglobulin M autoantibodies that agglutinate erythrocytes primarily at 4°C. Binding of cold agglutinins to erythrocytes takes place in acral parts of the circulation, with the immunoglobulin M cold agglutinin antibody activating the classical pathway of complement. Upon return to warmer parts of the circulation, antibodies dissociate from the cell surface, but C3b remains bound to the erythrocytes, causing extravascular hemolysis.35 C3b is eventually cleaved from surviving erythrocytes, leading to a high number of C3d-coated erythrocytes.36
Transplantation-associated thrombotic microangiopathy
Transplantation-associated thrombotic microangiopathy (TA-TMA) is a life-threatening complication of allogeneic hematopoietic cell transplantation.37 It manifests with the clinical triad of TMA: thrombocytopenia, microangiopathic hemolytic anemia, and organ damage (primarily involving the kidneys or central nervous system).38 However, diagnosis remains complex because of the high incidence of cytopenias and organ dysfunction in hematopoietic cell transplantation, as well as the lack of sensitivity of current diagnostic criteria.39 Recently proposed severity criteria have incorporated a rough marker of complement activation (soluble C5b-9), aiming to facilitate early diagnosis and treatment.40 Indeed, accumulating genetic and functional data suggest increased complement activation and possible genetic predisposition in both the adult and pediatric TA-TMA populations.41-43 In this complex setting of predisposing endothelial injury syndromes, other markers of endothelial damage, such as neutrophil extracellular traps, have also been found to be increased and warrant further study.41,44 In this context, MASP-2 has emerged as a potential therapeutic target based on its involvement in the complement pathway and direct interactions with the coagulation system.45,46 Because in vitro data on MASP-2 levels in TA-TMA are limited, they do not support its use as a marker in this setting.47 Therefore, the potential role of the lectin pathway in TA-TMA must be further investigated.
Complement inhibition in hematologic diseases
PNH
Since its introduction in 2007, eculizumab has become the standard of care in hemolytic PNH.48,49 Eculizumab is an anti-C5 humanized monoclonal antibody that binds to C5, preventing its cleavage into C5a and C5b24,50 (Figure 3). Thus, eculizumab disables the terminal complement pathway and inhibits MAC-mediated intravascular hemolysis, with all its clinical consequences.25,51 Two large international studies showed that eculizumab leads to hemoglobin stabilization and reduction of erythrocyte transfusion, with significant improvement in disease symptoms.26,27 Furthermore, eculizumab also markedly reduces thromboembolic risk in PNH28; all these clinical effects are retained in the long term with maintenance treatment,52 with no emergence of safety concerns (including infectious risk) so far.53 Available data suggest that the overall survival rate with eculizumab is as high as 95% at 5 years,54,55 which is much better compared with natural history.56 Nevertheless, efforts have been made to meet unmet clinical needs, starting with residual anemia during eculizumab treatment.57 Indeed, a recent real-life study exploiting a tentative response classification showed that, of 160 patients with PNH, only ∼20% achieved hemoglobin normalization.58 Recently, several novel anticomplement agents were investigated in PNH, addressing different unmet clinical needs.
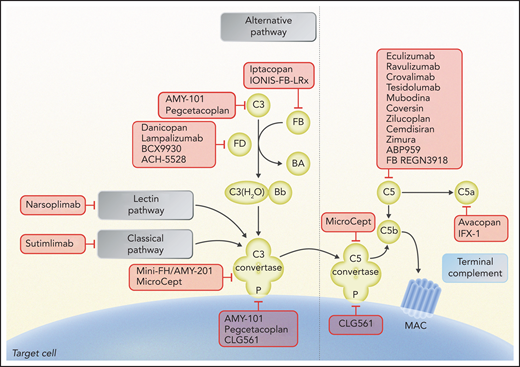
Schematic of complement activation, highlighting potential therapeutic targets under study. Complement inhibitors are summarized according to their target and the step of the complement pathway involved. Eculizumab, ravulizumab, crovalimab, tesidolumab, mubodina, coversin, zilucoplan, cemdisiran, zimura, ABP959, and REGN3918 inhibit C5; pegcetacoplan and AMY-101 inhibit C3 and C3 convertase activity; mini-FH/AMY-201 inhibits alternative pathway C3 convertase; iptacopan and IONIS-FB-LRx inhibit factor B; danicopan, lampalizumab, BCX9930, and ACH-5528 inhibit factor D; CLG561, pegcetacoplan, and AMY-101 inhibit properdin (P); sutimlimab inhibits C1s of the classical pathway; narsoplimab inhibits MASP-2 of the lectin pathway; mirococept inhibits C3 and C5 convertases; and avacopan inhibits C5a receptor and IFX-1 C5a. Professional illustration by Somersault 18:24.
Schematic of complement activation, highlighting potential therapeutic targets under study. Complement inhibitors are summarized according to their target and the step of the complement pathway involved. Eculizumab, ravulizumab, crovalimab, tesidolumab, mubodina, coversin, zilucoplan, cemdisiran, zimura, ABP959, and REGN3918 inhibit C5; pegcetacoplan and AMY-101 inhibit C3 and C3 convertase activity; mini-FH/AMY-201 inhibits alternative pathway C3 convertase; iptacopan and IONIS-FB-LRx inhibit factor B; danicopan, lampalizumab, BCX9930, and ACH-5528 inhibit factor D; CLG561, pegcetacoplan, and AMY-101 inhibit properdin (P); sutimlimab inhibits C1s of the classical pathway; narsoplimab inhibits MASP-2 of the lectin pathway; mirococept inhibits C3 and C5 convertases; and avacopan inhibits C5a receptor and IFX-1 C5a. Professional illustration by Somersault 18:24.
Development of second-generation complement inhibitors has occurred on 2 independent fronts. The first is optimization of anti-C5 therapy. Although more intriguing strategies have been attempted (eg, small interfering RNA59 and small peptide inhibitors60), advances have come from 2 long-acting monoclonal antibodies, ravulizumab and crovalimab. Ravulizumab is a derivative of eculizumab, from which it differs in 4 amino acid substitutions in complementarity-determining and Fc regions. These changes result in an extended half-life because of enhanced endosomal dissociation of C5 and recycling to the vascular compartment of the antibody through the neonatal Fc receptor. Ravulizumab was investigated in 2 large phase 3 studies enrolling untreated61 or eculizumab-treated patients with PNH,62 respectively. Ravulizumab was administered IV at 8-week dosing intervals,63 showing noninferiority to eculizumab with regard to lactate dehydrogenase (LDH) change or normalization, transfusion avoidance, breakthrough hemolysis, hemoglobin stabilization, and patient-reported outcomes.61,62 Ravulizumab was approved by the US Food and Drug Administration (FDA) and the European Medicines Agency in 2020. Crovalimab is another long-acting anti-C5 monoclonal antibody developed through a sequential monoclonal antibody recycling technology; given its high solubility, it can be delivered subcutaneously in small volumes.64 Crovalimab was tested in a 3-part open-label adaptive phase 1/2 trial investigating safety, pharmacokinetics, pharmacodynamics, and exploratory efficacy in healthy volunteers (part 1), as well as in complement blockade–naive (part 2) and C5 inhibitor–treated (part 3) patients.65 Once administered subcutaneously every month, crovalimab resulted in sustained inhibition of the complement terminal pathway, with adequate control of intravascular hemolysis, similar to other anti-C5 agents.65 Safety profile was excellent, although drug-target-drug complexes resulted in self-limiting autoimmune-like symptoms in a limited number of patients switching from eculizumab to crovalimab.65 Notably, the C5 epitope recognized by crovalimab is different from that bound by eculizumab; thus, crovalimab is pharmacologically effective even in patients carrying the C5 polymorphism, which accounts for lack of response in ∼3% of the Japanese population.66 Crovalimab is now under investigation in 2 large phase 3 studies enrolling untreated (COMMODORE 1; registered at www.clinicaltrials.gov as #NCT04432584) and eculizumab-treated (COMMODORE 2; #NCT04434092) patients with PNH, as summarized in Table 1.
The second front of development exploits a new strategy of inhibition that targets the early phases of complement activation.67 Pioneering preclinical work in PNH started with the first description of C3-mediated extravascular hemolysis.29,68 Indeed, the clinical development of proximal complement inhibitors for PNH found its rationale in the observation that the interception of the alternative pathway at the level of C3, or more upstream in the cascade, prevents C3 opsonization and MAC-mediated lysis in vitro.69,70 Different classes of compounds may lead to similar results in vitro; results are highly comparable, because the same experimental model was used, even if a formal head-to-head comparison was not shown. Indeed, engineered recombinant proteins (eg, complement factor H–related proteins TT3071 and mini-H72), the C3 inhibitor compstatin,73,74 and inhibitors of complement factor B75 and complement factor D76,77 were all equally effective in abolishing hemolysis while preventing C3 opsonization. Clinical development of the first proximal inhibitor investigated in vitro for PNH, TT30, was halted for pharmacokinetic reasons (despite proof of biologic efficacy78), but several proximal complement inhibitors have proceeded in clinical development, and they have been proven remarkably effective in recent clinical trials.57,79
The first PNH program exploiting compstatin was announced by Amyndas in 2012,73,74 using the third-generation compstatin analog AMY-101. In parallel, starting with the second-generation compstatin analog APL-180 (also known as POT-4), Apellis investigated a pegylated, long-acting version named APL-2 (now pegcetacoplan). After encouraging results in a proof-of-concept phase 1b study showing increased hemoglobin in 6 patients with PNH who had poor response to eculizumab,81 safety and efficacy of pegcetacoplan were investigated in a phase 3 open-label randomized study enrolling adult patients with PNH with hemoglobin <10.5 g/dL receiving eculizumab therapy (PEGASUS).82 After a 4-week run-in period with combination treatment, patients were randomly assigned to receive either subcutaneous pegcetacoplan monotherapy (n = 41) or IV eculizumab (n = 39). At week 16, pegcetacoplan was superior to eculizumab in hemoglobin change from baseline, with an adjusted mean treatment difference of 3.84 g/dL (P < .0001). Transfusion avoidance was higher, whereas noninferiority was demonstrated for absolute reticulocyte count, but not for LDH. Safety was acceptable, because most common treatment-emergent adverse events in pegcetacoplan and eculizumab groups, respectively, were injection site reactions, diarrhea, headache, and fatigue. There was no difference in infectious events between treatment arms; no cases of meningitis or thromboembolic events were observed during the 16-week study period. Breakthrough hemolysis rate was 10% in the pegcetacoplan arm at 16 weeks; 3 of 41 patients in the pegcetacoplan arm discontinued the study prematurely because of breakthrough hemolysis. Thus, pegcetacoplan in monotherapy was able to improve hematologic response in poor responders to eculizumab by preventing C3-mediated extravascular hemolysis while controlling MAC-mediated intravascular hemolysis at an acceptable level.82 These data were confirmed in a long-term (48 weeks) analysis conducted during open-label treatment with pegcetacoplan; patients randomly assigned to pegcetacoplan maintained high hemoglobin levels. In addition, patients initially randomly assigned to eculizumab who switched to pegcetacoplan during the open-label period demonstrated a significant improvement (2.9 g/dL) in hemoglobin levels compared with pretreatment. The rate of transfusion independence at 48 weeks was 73% in those initially randomly assigned to pegcetacoplan and 72% in those who switched from eculizumab to pegcetacoplan. The safety profile remained favorable, with no cases of meningitis; 1 death occurred as a result of COVID-19. Overall, 13 patients (15%) discontinued pegcetacoplan, 6 of whom because of breakthrough hemolysis and 1 because of mesenteric ischemia.83 Pegcetacoplan has just been approved by the FDA for patients with PNH who are switching from an anti-C5 treatment or are treatment naive.
The first-in-class factor D inhibitor danicopan, initially developed by Achillion, was investigated in 2 open-label single-arm phase 2 studies. The first enrolled 10 treatment-naive patients with PNH, who received danicopan orally as monotherapy at escalating doses (100-200 mg thrice daily).84 Eight of 10 patients completed the treatment (1 discontinued because of a serious adverse event and another because of personal reasons unrelated to safety), and all reached primary end point assessment. The primary end point was achieved, because danicopan led to significant LDH reduction (P < .001). Danicopan also resulted in a meaningful improvement of hemoglobin levels, with 1.1 and 1.7 g/dL increases from baseline at day 28 and day 84, respectively (both P < .005). The most common adverse events were headache and upper respiratory tract infection. Thus, monotherapy danicopan inhibits (but in most patients does not abolish) MAC-mediated intravascular hemolysis, with no evidence of C3-mediated extravascular hemolysis and clinically meaningful improvement of hemoglobin levels.84 In a parallel phase 2 study enrolling poor responders to eculizumab, danicopan was administered as add-on treatment at an oral dose of 100 to 200 thrice daily to 12 patients with PNH who remained transfusion dependent despite eculizumab treatment.85 In 11 patients evaluable for response (1 patient discontinued early because of a serious adverse event unlikely related to danicopan), at week 24, the addition of danicopan led to a mean hemoglobin increase of 2.4 g/dL. During the 24-week study period, only 1 transfusion (2 units) was administered to 1 patient, compared with the 34 transfusions (58 units) administered to 10 patients in the 12 weeks before enrollment. Headache was the most common adverse event. Thus, as add-on treatment, danicopan resulted in prevention of C3-mediated extravascular hemolysis and subsequent hemoglobin improvement in poor responders to eculizumab.85 Danicopan is now under investigation as add-on therapy in a phase 3 randomized study of patients with PNH with poor response to eculizumab or ravulizumab (#NCT04469465). At the same time, an analog of danicopan was developed (ACH-5528 or ALXN-2050) and is currently being investigated as monotherapy (#NCT04170023).
Another factor D inhibitor named BCX9930 has been developed by Biocryst; phase 1b data were recently reported. In 6 patients with PNH with residual anemia despite anti-C5 treatment, BCX9930 administered at doses up to 500 mg twice daily orally was safe and generally well tolerated, leading to significant improvement of hemoglobin levels and hemolytic biomarkers.86 BCX9930 was also investigated at different doses in 10 treatment-naive patients with PNH; the treatment in general seemed safe, although 1 case of fatal disseminated varicella zoster infection raises the need for additional data. The fully therapeutic dose of ≥400 mg twice daily resulted in transfusion independence in all patients, with a mean improvement in hemoglobin level of 3.9 g/dL.87 Based on these data, a phase 3 program has been announced.
The first-in-class factor B inhibitor iptacopan was investigated in an open-label phase 2 study enrolling patients with PNH with signs of active hemolysis during eculizumab treatment.88 The study was conceived initially as an add-on treatment, with iptacopan administered at a dose of 200 mg twice daily orally on top of standard-of-care eculizumab; the primary end point was change in LDH level at 13 weeks. After this initial study period, a subsequent extension study offered to all patients reaching the primary end point allowed changes to standard-of-care treatment, including possible discontinuation. Data are available for the first 10 patients; all received iptacopan as per protocol, with no treatment discontinuation or treatment-related serious adverse events. Treatment was well tolerated, without safety concerns. The primary end point of the study was achieved, with marked LDH reduction in all patients (P = .006). Remarkably, 8 of 10 patients achieved full normalization of hemoglobin levels, with mean hemoglobin level increasing from 9.77 ± 10.5 g/dL at baseline to 12.63 ± 1.85 g/dL at 13 weeks (P < .001). Iptacopan also led to significant reduction of bilirubin and absolute reticulocyte count; furthermore, C3 deposition of PNH erythrocytes became undetectable, and the size of the PNH erythrocyte population markedly increased. These data are consistent with a full prevention of C3-mediated extravascular hemolysis, associated with improved inhibition of MAC-mediated intravascular hemolysis.88 In the subsequent extension study, all the positive effects were retained, even in 7 patients who discontinued eculizumab and continued iptacopan as monotherapy. In these patients switching to iptacopan as monotherapy, no changes in LDH, hemoglobin, or any other biomarker of hemolysis (including C3 deposition and PNH erythrocyte population) were observed after discontinuation of eculizumab. Thus, iptacopan, either as add-on or single-agent treatment, led to full prevention of C3-mediated extravascular hemolysis and better control of MAC-mediated intravascular hemolysis in poor responders to eculizumab.88 Preliminary data on iptacopan monotherapy in treatment-naive patients with PNH were recently reported; in 13 patients who were randomly assigned to receive different doses of iptacopan, control of intravascular hemolysis was achieved, with improvement of hemolytic biomarkers and hemoglobin. The highest effect was observed in patients receiving the dose of 200 mg twice daily, setting this as the standard dose for additional studies.89 Indeed, iptacopan is now under investigation in a large phase 3 multicenter trial enrolling patients with PNH with suboptimal hematologic response to standard-of-care anti-C5 treatment (#NCT04558918).
CAD
Symptomatic treatment of CAD includes avoidance of cold temperatures if possible, especially in acral parts of the circulation. Pharmacologic treatment involves rituximab as the best first-line therapy, leading to remission (median duration, 1 year) in >50% of patients.90 In relapsed cases, alternative treatment options have been described: subsequent rituximab therapy; fludarabine/rituximab combination therapy, with a 75% response rate (median duration, >5 years) but significantly more toxicity90; rituximab and bendamustine combination therapy; and bortezomib-based therapy.91
Current updates of CAD management have focused on complement inhibition, after initial proof that classical pathway inhibition prevents generation of anaphylatoxins driven by cold agglutinin.35 Sutimlimab is a first-in-class humanized monoclonal antibody that binds to C1s and inhibits classical complement activation (Figure 3). A phase 1b trial of sutimlimab demonstrated that weekly IV dosing for 4 weeks followed by biweekly dosing thereafter rapidly aborted C1s complement-mediated hemolysis and significantly increased hemoglobin levels, precluding the need for erythrocyte transfusions.92 All patients responded to sutimlimab within a few weeks, with a median rise in hemoglobin of almost 4 g/dL. It should be noted that sutimlimab does not affect the production of cold agglutinins or their binding to erythrocyte antigens; therefore, patients with CAD may still experience acrocyanosis. The randomized CADENZA and the open-label CARDINAL trials of sutimlimab have shown improvements in hemoglobin levels, hemolysis markers, fatigue, and quality-of-life indices.93-95 Serious (including bacterial) infections were reported, but no meningococcal infections were identified. Another complement inhibitor that targets C3, pegcetacoplan (Figure 3), was also studied in a phase 2 trial of autoimmune hemolytic anemia. The interim analysis of 10 patients with CAD showed improvements in hemoglobin, hemolytic markers, and fatigues scores.96 Therefore, targeted complement treatment is expected to become standard of care for CAD.
TA-TMA
In the preeculizumab era, standard of care for TA-TMA involved cessation of calcineurin or mammalian target of rapamycin inhibitors, supportive treatment, corticosteroids, and sometimes plasma exchange or rituximab according to each center’s policy. Over the past years, eculizumab has been increasingly used to treat both adult and pediatric patients with TA-TMA.97-101 Despite high response rates to eculizumab treatment that have reached 93% in some studies, overall survival has remained low (∼30%) in early reports from the adult population.99,100 Importantly, Jodele et al101 achieved an increased 1-year survival of 66% in 64 pediatric eculizumab-treated patients, compared with 17% in a historical control group.
Regarding novel complement inhibitors, a C5 inhibitor, coversin, was successfully used in a patient with TA-TMA with a C5 variant that caused resistance to eculizumab treatment.102 Interestingly, a phase 2 single-arm open-label study of the MASP-2 inhibitor narsoplimab (Figure 3) in 19 patients with TA-TMA also reported increased median overall survival compared with a historical control of conventional treatment (347 vs 21 days from TA-TMA diagnosis).103 The drug is currently under priority review by the FDA. It should also be noted that the long-acting C5 inhibitor approved for PNH, ravulizumab, is being studied in a phase 3 trial of adult (#NCT04543591) and pediatric (#NCT04557735) patients with TA-TMA (#NCT02946463). Lastly, coversin or nomacopan, a C5 inhibitor that also blocks leukotriene B4, is being evaluated in a 2-part phase 3 trial in pediatric TA-TMA (#NCT04784455).
Hematologic entities under investigation for complement inhibition
Sickle cell disease
Accumulating evidence points toward a key role of complement in life-threatening complications of sickle cell disease, such as delayed hemolytic transfusion reaction (DHTR).104 To this end, a number of case reports and series have documented the use of eculizumab as successful salvage therapy for DHTR.105-109 In line with these data, 2020 guidelines by the American Society of Hematology have recommended complement inhibition in patients with DHTR and ongoing hyperhemolysis.110
Immune thrombocytopenia
The pathophysiology of immune thrombocytopenia (ITP) is considered multifactorial, with complement being activated by autoantibodies bound to the platelet surface.111,112 Sutimlimab, the C1s inhibitor under advanced clinical development, has induced rapid and durable responses in patients with chronic ITP who were refractory to multiple prior therapies.113 In an era with several novel agents under development for ITP,114 complement inhibition might be part of the future armamentarium against the disease.
Other complement-related disorders of potential interest to hematologists
The role of complement as a therapeutic target is currently being investigated in additional entities that may be of interest to the broader hematology and immunotherapy community. First, atypical HUS is the prototypic model of complement activation in TMA,115 and the standard anti-C5 agents eculizumab and ravulizumab are approved this indication. With a focus primarily on hematologic diseases, this review does not address atypical HUS in detail. In addition, catastrophic antiphospholipid syndrome is characterized by excessive complement activation in patients with underlying complement-related genetic variants.116 As a result, several small reports have shown successful outcomes with eculizumab, and prospective clinical trials are still expected in this field.117 Furthermore, complement also plays a crucial part in hyperinflammation and may contribute to interleukin-6–mediated cytokine release syndrome.118 This involvement has been studied extensively in patients with COVID-19, with functional and genetic evidence of complement activation.119-122 Prompted by promising results of complement inhibitors in case series, clinical trials are evaluating C3 and C5 inhibitors in COVID-19 treatment.123-125
Discussion: emerging questions with proximal complement inhibitors
Our state-of-the-art review reveals advances and challenges in the continuously evolving field of targeted complement therapy in hematologic diseases. Among them, PNH remains the prototypic model of complement activation and inhibition, and recent data have raised novel questions in the field, paving the way for future changes in the paradigm of PNH treatment. Indeed, the first proximal inhibitor, pegcetacoplan, was recently approved by the FDA, and oral factor B and factor D inhibitors are under investigation in phase 3 registration trials.84,88,126 Although all proximal inhibitors can equally provide hematologic benefits by preventing extravascular hemolysis, their sustained effects on intravascular hemolysis as monotherapies is a matter of debate. Knowledge of complement biology and preclinical data suggest that if the proximal pathway is adequately blocked, the terminal pathway will be preempted as well. Indeed, this concept has been confirmed in vivo, but not all proximal targets are equivalent, and not all inhibitors are the same. For instance, C3 is a high-concentration (and high turnover) plasma protein serving as a proenzyme and its own substrate, whereas factor B and factor D are, respectively, a proenzyme and an enzyme with lower plasma concentration. Furthermore, C3 is a key molecule irrespective of the specific complement pathway initially triggered, whereas factor B and factor D are involved only in the alternative (and in the amplification) pathway. In addition, direct factor B activation by plasma protease, bypassing factor D, has been described.127 Because hemolysis of PNH in vivo is due to the alternative pathway, C3, factor B, and factor D seem equally excellent as therapeutic targets, with the caveat of possible factor D bypass (which remains to be demonstrated in humans and in PNH). However, even if targets are mechanistically equivalent, individual inhibitors are not, as shown by available data. For instance, pharmacodynamic and pharmacokinetic properties of each inhibitor shape their individual efficacy and safety, because the risk of transient leaks in therapeutic inhibition may lead to reappearance of hemolysis. This is even more important because the actual mass of erythrocytes susceptible to hemolysis largely increases on proximal inhibitors (the proportion of PNH erythrocytes may exceed 90%, which is the strongest proof of their remarkable efficacy), eventually generating a new form of PNH, the clinical course and possible complications of which remain to be elucidated. Available data suggest even such large PNH erythrocyte masses are not necessarily associated with meaningful breakthrough hemolysis per se, but clinically relevant breakthrough hemolysis may occur in cases of subtherapeutic plasma levels of the inhibitor (described with both pegcetacoplan and danicopan84,126) and/or in cases of any complement-amplifying condition that may overcome even therapeutic plasma levels of the inhibitor (Figure 2). Although breakthrough hemolysis even on proximal inhibitors seems to be self-limiting in most cases, in some circumstances hemolysis can be massive; in the pegcetacoplan study, this occasionally led to discontinuation of the proximal inhibitor, suggesting that specific therapeutic interventions may be necessary, including an extra dose of the proximal inhibitor (eg, dose adjustment was allowed in the pegcetacoplan study) or even rescue treatment with a terminal inhibitor. Furthermore, adherence to treatment may be affected by administration route and dose frequency, as well as by patient compliance; therefore, oral treatment seems more convenient than subcutaneous treatment, provided that pharmacodynamic and pharmacokinetic properties allow some flexibility in daily schedule and at least 1 missed dose without leaks of inhibition (as apparently seen with iptacopan, but not with danicopan). Long-term data on proximal inhibitors are needed to better understand how to best use them, maximizing the advantage of their powerful efficacy while limiting their potential risks. It is very likely that proximal inhibitors will represent the next standard of care for PNH, but it remains to be confirmed that concomitant anti-C5 treatment is not necessary to maximize the hematologic response or prevent/treat breakthrough hemolysis in some patients. Because complement is involved in different mechanisms of surveillance against infectious agents as well as against autoimmune damages and possibly cancer, future studies will rule out any increased risk of these complications during long-term use of proximal complement inhibitors.128
Beyond PNH, in addition to the well-established use of C5 inhibitors for atypical HUS, these novel strategies of complement inhibition have also shown efficacy and safety in CAD, primarily with the C1s inhibitor of the classical complement pathway sutimlimab, but also with pegcetacoplan. Furthermore, inhibition of the lectin pathway with narsoplimab is being investigated in TA-TMA, a condition in which terminal inhibitors have been used and are now being systematically investigated. With this revolution of next-generation complement therapeutics, additional hematologic entities, such as DHTR or ITP, might also benefit from further study of the safety and efficacy of complement inhibitors. Lastly, other complement-related disorders of potential interest to hematologists, including catastrophic antiphospholipid syndrome and hyperinflammation, may advance our understanding of complement diagnostics and therapeutics.
Acknowledgments
Given the broad scope of this review, the authors often refer to specialized review articles rather than primary literature, and they have been able to include only selected examples of original work in the field. Therefore, the authors thank colleagues who are not specifically cited for their contribution and their understanding.
E.G. is supported by the ASH Global Research Award 2020.
Authorship
Contribution: E.G. researched the literature and contributed to discussion of the content; R.P.d.L. and A.M.R. selected the publicly available data from the original investigations quoted in the manuscript and contributed to discussion of the content; and all authors wrote the text and edited the manuscript before submission.
Conflict-of-interest disclosure: E.G. has consulted for Amyndas, Alexion, and Omeros Pharmaceuticals, Inc. R.P.d.L. has received research support from Alexion, Novartis, Pfizer, and Amgen; received lecture fees from Alexion, Novartis, Pfizer, and Apellis/SOBI; served as member of advisory/investigator board for Alexion, Apellis/SOBI, Biocryst, Novartis, Roche, and Samsung; and served as consultant for Alexion, Novartis, and Apellis. A.M.R. has received research support from Alexion, Novartis, Alnylam, and Rapharma; received lecture fees from Alexion, Novartis, Pfizer, and Apellis/SOBI; served as member of advisory/investigator board for Alexion, Achillion, Apellis/SOBI, Biocryst, Novartis, Roche, Samsung, and Sanofi; and served as consultant for Amyndas, Novartis, and Omeros.
Correspondence: Eleni Gavriilaki, Hematology Department-BMT Unit, G. Papanicolaou Hospital, Exochi, 57010 Thessaloniki, Greece; e-mail: elenicelli@yahoo.gr.


