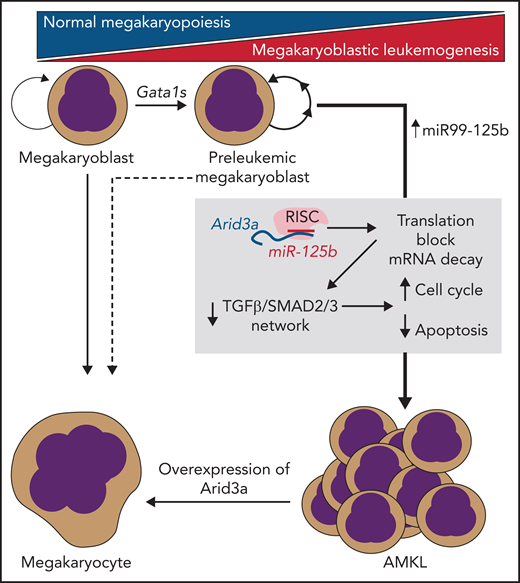
In this issue of Blood, 1 provide new insight into how trisomy 21 and truncating mutations in the Gata1 transcription factor collaborate to generate acute megakaryoblastic leukemia (AMKL).
AMKL is an aggressive subtype of acute myeloid leukemia that is believed to originate within malignant megakaryoblasts in the bone marrow. Although N-terminal truncating mutations in Gata1 (leading to production of the dominant-negative Gata1s transcription factor) have been shown to be a near-universal feature of AMKL leukemogenesis, expression of Gata1s alone seems to be insufficient to cause fulminant AMKL. Although several mutations and deregulated pathways, such as CHAF1B,2RUNX1, ETS2, and ERG,3 have been shown to participate in this process, the strong clinical correlation between AMKL and patients with Down syndrome suggested a mechanism intrinsic to chromosome 21 overexpression that had not been fully elucidated. Building on prior work from the Klusmann group, who demonstrated a key role for the microRNA (miRNA) cluster miR-99-125b found on chromosome 21 in AMKL pathogenesis,4,5 the investigators provide compelling evidence for miR-125b repression of Arid3a as the important complementary “hit” in leukemia progression.
miRNAs are short noncoding RNAs that posttranscriptionally control the expression of target RNAs. Although the biogenesis and control of miRNA expression are complex (see Bartel6 and Gebert and MacRae7 for more details), the central theory of how miRNAs regulate gene expression is through licensing of Argonaute (Ago) proteins to the 3' untranslated region of target RNAs. The miRNA-Ago complex (RNA-induced silencing complex [RISC]) induces translation inhibition and messenger RNA (mRNA) decay. This evolutionarily conserved form of posttranscriptional regulation by miRNAs has been shown to be important in a variety of biologic contexts, including development and oncogenesis.7
In their study, Alejo-Valle et al use a CRISPR-based model of Gata1s-driven AMKL preleukemia to screen for the synergistic leukemic activity of each member of the miR99-125b cluster, which demonstrated that miR-125b was the putative driver miRNA among the cluster members. Following from this result, the group performed an elegant series of experiments to identify the transcription factor Arid3a as the most likely functionally relevant candidate target RNA that was being silenced via miR-125b. Consistent with their screen, targeted disruption of Arid3a could participate with Gata1s to induce AMKL in vivo, even in the absence of miR-125b, and overexpression of this factor could lead to rapid differentiation and proliferation block of AMKL cell lines and AMKL patient samples (see figure). Further studies aimed at understanding the mechanism of Arid3a’s role in leukemogenesis demonstrated that it may play roles in Gata1-induced differentiation networks, as well as in cell cycle arrest and apoptosis induced by transforming growth factor-β (TGF-β)–SMAD2/3. Taken together, Arid3a appears to be an essential player in normal megakaryopoiesis and a key target in AMKL pathogenesis.
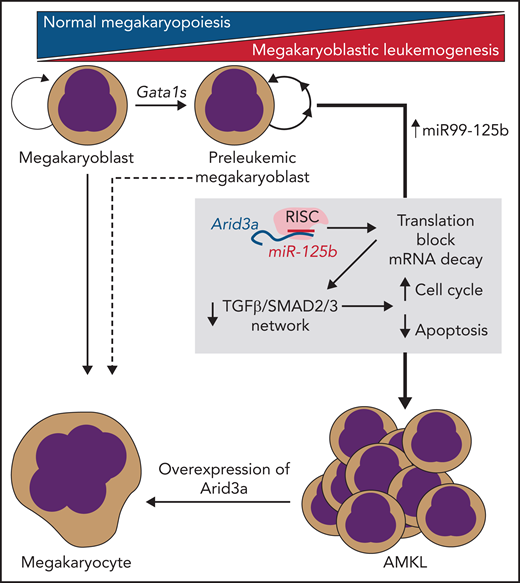
miR-125b/RISC–mediated translational blockade and mRNA decay of Arid3a are necessary and sufficient to transform Gata1s-mutant preleukemic megakaryoblasts into fulminant AMKL. This process is mediated via disruption of TGF-β/SMAD2/3–induced transcription networks by loss of Arid3a.
miR-125b/RISC–mediated translational blockade and mRNA decay of Arid3a are necessary and sufficient to transform Gata1s-mutant preleukemic megakaryoblasts into fulminant AMKL. This process is mediated via disruption of TGF-β/SMAD2/3–induced transcription networks by loss of Arid3a.
Their work represents an important milestone in our understanding of AMKL biology, while setting the stage for multiple lines of further inquiry. The robust multidimensional approach taken by the investigators provides a clear role for a miR-125b-Arid3a axis in promoting the preleukemia-leukemia transition in Gata1s-mutant megakaryoblasts. This result, in particular, helps to explain why overexpression of chromosome 21 in Down syndrome leads to such a dramatic increase in the rate of AMKL development. Second, the systematic analysis provides some of the strongest evidence for miRNA-mediated leukemogenesis to date. Although the complex biology and often pleiotropic effects of these dynamic molecules are still being worked out, the study serves as a potent example of how the “noncoding” genome can lead to substantial phenotypic changes in the cell. Third, the identification of Arid3a’s previously unknown role in megakaryocytic differentiation, particularly as a partner to Gata1 and Smad2/3, extends the repertoire of functions that this ubiquitous transcription factor plays in promoting normal hematopoiesis. With these points in mind, a number of key questions arise. First, the results of this study beg the important questions of how the miR99-125b cluster is regulated throughout the hematopoietic hierarchy, as well as how each of the miRNAs encoded in this polycistronic RNA are “tuned” to meet the requirements of various hematopoietic stem and progenitor cell compartments. Second, what distinct epigenetic signatures are facilitated via Arid3a’s collaboration with Gata1 and Smad2/3, and why is Arid3a necessary to induce transcriptomes compatible with megakaryocytic differentiation? These latter questions, in particular, could open the door to exploring pharmacologic intervention to “correct” deregulated epigenetic signatures in AMKL megakaryoblasts as an adjuvant to current standard of care.
In summary, the study by Alejo-Valle and colleagues offers novel insight into the molecular pathogenesis of AMKL. By using multi-omics and forward and reverse genetics, the investigators made a compelling and robust case for the role of miR-125b and, subsequently, deregulated Arid3a as central to Gata1s-driven leukemogenesis. Importantly, this work has also opened the door to multiple new areas of inquiry in normal megakaryopoiesis and AMKL pathogenesis, as well as solidified the potent role that miRNAs can play in shaping cellular fates. This study represents a significant step in our understanding of the molecular mechanisms of megakaryoblastic leukemia and will hopefully translate into better more targeted therapeutics for treating these aggressive malignancies.
Conflict-of-interest disclosure: U.S. has received research support and personal fees from Bayer Healthcare and Aileron Therapeutics; personal fees from Celgene, Pieris Pharmaceuticals, Trillium Therapeutics, Novartis, and Vor Biopharma; research support from GlaxoSmithKline; and personal fees and involvement as scientific cofounder and member of the board of directors of Stelexis Therapeutics. J.C.W. declares no competing financial interests.