In this issue of Blood, report that downregulation of MS4A3 contributes to the persistence, progression, and drug resistance of chronic myeloid leukemia (CML) to the BCR-ABL inhibitor imatinib.1 Thus, enhancing MS4A3 levels provides a potential therapeutic strategy in treating CML.
CML arises from abnormal hematopoietic stem cells called leukemia stem cells (LSCs). These LSCs carry a BCR-ABL1 fusion gene that encodes a constitutively active ABL kinase triggering a wide range of signaling pathways. These signaling pathways favor the expansion and differentiation of leukemia stem and progenitor cells (LSPCs), leading to excessive production of myeloid cells.2 Without effective treatment, chronic-phase (CP) CML eventually evolves to blast-phase (BP) CML, characterized by increased immature myeloid blasts and the acquisition of additional genetic abnormalities.3 Before the advent of targeted therapy, the median time for this malignant transformation was ∼3 to 4 years.2 Prognosis is dismal after transformation to acute leukemia, also called blast crisis.
The nature of BCR-ABL1 fusion protein as a tyrosine kinase renders it an ideal therapeutic target. The first “molecular bullet,” imatinib, competes with the ATP-binding site in ABL, thus inhibiting kinase activity. Subsequently, the US Food and Drug Administration has approved 5 additional tyrosine kinase inhibitors (TKIs) for CML. The TKIs have transformed CML treatment, turning a fatal disease to a chronic condition.2 Imatinib exhibits great efficacy in suppressing CP-CMLs, with most of the patients achieving a remarkable reduction of BCR-ABL1+ cells and some with deep molecular remissions.2,4 However, BCR-ABL TKIs are less effective in treating BP-CMLs. Both BCR-ABL1 kinase-dependent and -independent mechanisms account for TKI resistance, including primary resistance (failure to achieve an optimal response) and relapse after achieving a response. Some patients develop resistance owing to on-target mutations in the ABL kinase, rendering them insensitive to TKIs. The recently approved TKI, asciminib, is an allosteric inhibitor that targets the ABL1 myristoyl pocket, overcoming kinase domain resistance mutations. It will be important to see if new mutations will arise in the asciminib-treated patients and if asciminib combined with ATP site TKIs will achieve greater efficacy clinically.5 Furthermore, reactivation of BCR-ABL1 downstream signaling contributes to drug resistance. Mutations in transcriptional and epigenetic regulators also contribute to BCR-ABL1 kinase independent resistance. They are associated with a poor outcome and progression to blast crisis.6 Hence, a better understanding of CML resistance and treatment relapse should point to new strategies to cure this disease.
It has been shown that a pool of quiescent BCR-ABL1+ LSCs do not rely on BCR-ABL1 kinase activity to survive, thus are not eliminated by TKI treatment.7 These CML LSCs contribute to primary resistance, and their persistence despite effective BCR-ABL1 inhibition also contributes to relapse upon the cessation of TKI treatment. In this issue of Blood, Zhao et al demonstrate a previously unappreciated mechanism that governs the maintenance of CML quiescence and BP-CML by modulating cytokine signaling and cytokine-mediated differentiation (see figure).1 Through a comprehensive analysis of published CML transcriptomes, they identify that low expression of MS4A3, a member of the membrane-spanning 4-domains A (MS4A) family proteins, is a characteristic of LSC quiescence, primary TKI resistance, and BP-CML. MS4A3 expression is low in CD34+CD38− stem cells in both patients with normal bone marrow and patients with CML. Interestingly, although MS4A3 expression is upregulated in normal CD34+CD38+ and CD34−CD38− progenitor cells that correlate with differentiation,8,9 it remains low in CML LSPCs. Functionally, overexpression of MS4A3 reduced, whereas inducible MS4A3 knockdown increased, colony formation of CML CD34+ cells. Depletion of MS4A3 in vivo also augmented CML engraftment in xenotransplanted mice. This observation is confirmed in Ms4a3-deficient mice in a Bcr-Abl1 transgenic background. It is important to note that changes of MS4A3 expression in normal HSPCs or BCR-ABL1− CD34+ cells do not affect progenitor cell growth.
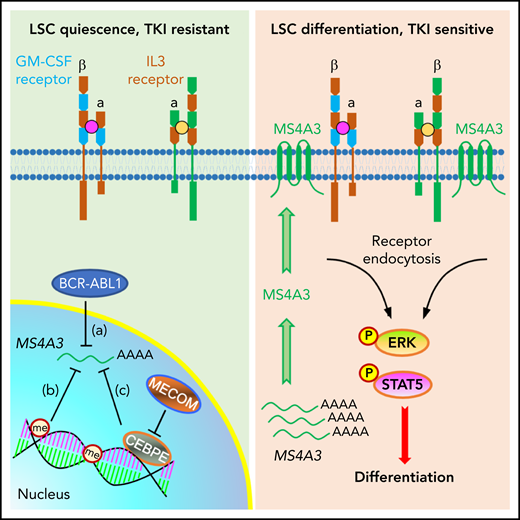
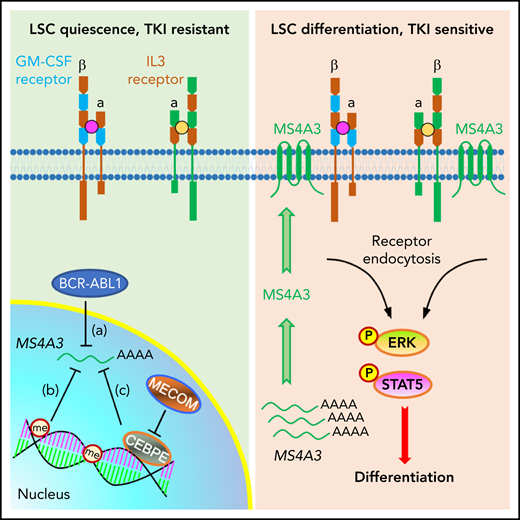
A working model depicting a role for MS4A3 in regulating CML LSC differentiation and TKI sensitivity, via modulating common βc receptor endocytosis and signaling. Left: in BCR-ABL1+ CML LSCs, BCR-ABL signaling (a), DNA and histone methylation (me) (b), as well as the MECOM/CEBPE axis (c) transcriptionally downregulate MS4A3, that encodes a tetraspan membrane protein. Low MS4A3 levels contribute to LSC quiescence, TKI resistance, and differentiation block in BP-CML. Right: MS4A3 levels are high in TKI-sensitive CML, or when exogenously delivered, they bind to the common βc for the GM-CSF and IL-3 receptors in the cell membrane, promote receptor endocytosis and activation of ERK and STAT5, thereby enhancing LSC differentiation.
A working model depicting a role for MS4A3 in regulating CML LSC differentiation and TKI sensitivity, via modulating common βc receptor endocytosis and signaling. Left: in BCR-ABL1+ CML LSCs, BCR-ABL signaling (a), DNA and histone methylation (me) (b), as well as the MECOM/CEBPE axis (c) transcriptionally downregulate MS4A3, that encodes a tetraspan membrane protein. Low MS4A3 levels contribute to LSC quiescence, TKI resistance, and differentiation block in BP-CML. Right: MS4A3 levels are high in TKI-sensitive CML, or when exogenously delivered, they bind to the common βc for the GM-CSF and IL-3 receptors in the cell membrane, promote receptor endocytosis and activation of ERK and STAT5, thereby enhancing LSC differentiation.
Having shown the importance of MS4A3 in CML LSPCs, the authors interrogated the signals that regulate MS4A3 expression. Interestingly, BCR-ABL1 reduces MS4A3 expression in primary CML, but in BP-CML, MS4A3 is downregulated and insensitive to imatinib treatment. They found that epigenetic regulation, including promoter hypermethylation and EZH2/PRC2 mediated-chromatin changes, as well as the MECOM/CEBPE axis control MS4A3 expression.
The authors next investigated the mechanisms by which MS4A3 regulates LSPC expansion. Using live cell imaging, they discovered that MS4A3 traffics between the plasma membrane and endosomes. MS4A3 depletion increased membrane-bound proteins, including the common β chain (βc) of the granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-3 (IL-3), and IL-5 receptors. MS4A3 enhanced CML LSPC differentiation by promoting endocytosis and activation of pERK and pSTAT5 emanating from the GM-CSF and IL-3 receptors. Encouraged by these findings, the authors explored the therapeutic potential of manipulating MS4A3 expression in drug-resistant CML LSPCs. Delivery of MS4A3 proteins using nanoparticles reduced CD34+CD38− LSCs, promoted their differentiation, and significantly reduced colony formation, attesting to its therapeutic potential.
This study sheds significant light into our understanding of how the BCR-ABL fusion protein leads to detrimental changes that promote CML progression, how some CML LSCs are able to maintain quiescence and insensitivity to cytokine-induced differentiation, and how residual CML LSCs persist and relapse after TKI discontinuation. The data in this work argue for a role for MS4A3 as a tumor suppressor that blocks cytokine signaling and cytokine-mediated differentiation in LSPCs. It also broadens our knowledge on MS4A family transmembrane proteins in regulating membrane receptor signaling.10 Based on this work, some interesting questions are worthy of further studies. Does MS4A3 play a role in other myeloid malignancies? Does MS4A3 anchor common βc receptors to specific membrane microdomains, such as a lipid raft or lysosomes that could affect receptor turnover? What are the additional targets of MS4A3? The novel finding in this work also points to elaborate regulation of MS4A3 level as a molecular switch to control the balance of LSC quiescence, expansion, and differentiation. How does BCR-ABL1 cause epigenetic changes and activate the MECOM/CEBPE axis? Further study is needed to devise new therapies to increase MS4A3 expression, and, when combined with existing TKIs, to eradicate CML LSPCs. It also indicates that targeted delivery of MS4A3 may drive deeper molecular responses in patients with CML on TKI therapies, thus providing novel therapeutic potential for eliminating CML LSPCs.
Conflict-of-interest disclosure: The authors declare no competing financial interests.