

In this issue of Blood, identify heterozygous missense variants clustered around the adenosine triphosphate (ATP)-binding pocket of caseinolytic peptidase B (CLPB) as a novel genetic cause of severe congenital neutropenia (SCN).1 Mechanistically, these CLPB variants impair mitochondrial function and granulopoiesis in a dominant-negative manner.
SCN is a heterogenous preleukemia bone marrow failure syndrome with mutations in >20 different genes reported to be causative for the disease.2 The most frequently mutated gene, ELANE, encodes for neutrophile elastase, a protein released by neutrophils to fight infections. Here, Warren et al identify 6 new heterozygous missense variants in the CLPB protein homolog (CLPB) in 10 unrelated patients with genetically unresolved SCN.
All variants are located in the evolutionarily conserved ATP-binding pocket on the CLPB protein, which is essential for the hydrolysis of ATP. How cellular ATP content regulates granulocytic differentiation of hematopoietic stem/progenitor cells (HSPCs) is unclear. To further investigate the role of CLPB in hematopoiesis, the authors used lentiviruses to overexpress wild type and 4 of the identified missense variants of CLPB in primary human hematopoietic stem/progenitor cells (HSPCs). They found that both wild-type and variant CLPB are localized in mitochondria, but only ATP-binding pocket CLPB variants induce mitochondrial dysfunction and decrease ATP production. Functionally, CLPB variants impair granulocytic differentiation and enhance apoptosis without affecting the cell cycle. Increased apoptosis of myeloid progenitors has been previously described as one of the causes of SCN.3 Interestingly, stress on the endoplasmic reticulum (ER), a common finding in myeloid progenitors in patients with SCN,4,5 was not a hallmark of CLPB-induced intracellular defects.
The authors investigated the mechanism by which faulty ATP hydrolysis alters granulopoiesis downstream of missense CLPB mutations and found that mutated CLPB acts in a dominant-negative fashion disrupting wild-type CLPB (see figure). Consistently, CLPB variant–overexpressing cells showed similar phenotypes as CLPB-null cells generated by CRISPR-Cas9 gene editing. These findings are also important from a therapeutic point of view, as they indicate that correction of mutated alleles by gene therapy is a rather promising treatment strategy for these patients. Instead, strategies involving complementation with wild-type protein will likely be ineffective, due to persistent inhibition of the mutant protein. Dominant-negative effects of mutated vs wild-type protein have also been recently described as important for the pathogenesis of SCN with SRP54 mutations.6
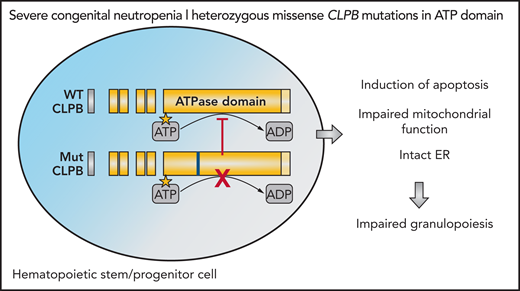
CLPB variants in genetically unresolved SCN. Heterozygous missense ATP-binding pocket variants in CLPB act in a dominant-negative manner and disrupt wild-type (WT) CLPB function. Expression of CLPB variant is associated with mitochondrial dysfunction, increased apoptosis, and impaired granulocytic differentiation in HSPCs. ADP, adenosine 5′-diphosphate; ATPase, adenosine triphosphatase; Mut, mutant.
CLPB variants in genetically unresolved SCN. Heterozygous missense ATP-binding pocket variants in CLPB act in a dominant-negative manner and disrupt wild-type (WT) CLPB function. Expression of CLPB variant is associated with mitochondrial dysfunction, increased apoptosis, and impaired granulocytic differentiation in HSPCs. ADP, adenosine 5′-diphosphate; ATPase, adenosine triphosphatase; Mut, mutant.
It is remarkable to see that mutations in genes encoding proteins with entirely different functions, intracellular localization, and activity lead to neutropenia. Most of them induce maturation defects of granulocytic differentiation in the bone marrow at the stage of granulocytic progenitors, that is, promyelocytes and myelocytes. For some inherited neutropenia syndromes, for example, neutropenia associated with mutations in the ELANE gene, patients have isolated neutropenia without any defects in other organs or blood cells.7 For other gene mutations, for example, SRP54, G6PC3, and JAGN1, severe neutropenia is often associated with other dysfunctions of the vascular, the gastrointestinal and/or the nervous system.2 The patients described by Warren et al also presented with maturation arrest of granulopoiesis in the bone marrow, which led to typical severe bacterial and fungal infections. Some of the patients had additional abnormalities including neurologic symptoms, cataracts, and other organ abnormalities. The patients were treated with granulocyte colony-stimulating factor, which is the standard treatment of the majority of SCN patients. SCN is a preleukemia syndrome, and one of the patients with mutant CLPB also developed acute myeloid leukemia (AML)/myelodysplastic syndrome (MDS). Because we previously described the high frequency of acquired cooperating mutations in the CSF3R and RUNX1 genes in SCN patients who developed overt MDS or AML,8 it would be interesting to test these mutations in CLPB-associated patients with SCN. Interestingly, a missense CLPB mutation was also detected in 1 patient with cyclic neutropenia (CyN), another inherited neutropenia syndrome with a 21-day period of cycling neutrophil counts. Approximately 90 percent of CyN patients harbor mutations in the ELANE gene, which is also the most commonly mutated gene in SCN. This study is the first to describe another mutation detectable in both SCN and CyN patients. Although CLPB has emerged as a gene to be sought in patients with genetically unresolved SCN, the contribution of the different CLPB variants to the disease requires further study. CLPB is expressed in the mitochondrial compartment of various tissues (eg, adult brain and neutrophils) and functions as a potent protein disaggregase and ATP-dependent chaperone.9 The biallelic composition of CLPB variants has been associated with a so-called CLPB syndrome, an autosomal-recessive disorder characterized by 3-methylglutaconic aciduria (3-MGA), cataracts, neurologic disease, and variable neutropenia.10,11 Studies in relatives from patients with CLPB syndrome reveal heterozygous carriers as clinically asymptomatic. However, the current study shows that heterozygote missense mutations in ATP-binding pocket positions are sufficient to induce severe neutropenia, but with no consistent nonhematologic findings.
In sum, heterozygous missense ATP-binding pocket CLPB variants are a new genetic abnormality in SCN. Different phenotypes are likely induced by CLPB variants dependent on the mutation’s position and the gene dosage. The data reported by Warren et al not only expand the diagnostic options for SCN patients, bringing 1 more genetic player into the diagnostic pool, but also provide novel information for a better understanding of the biology of granulopoiesis in general.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

