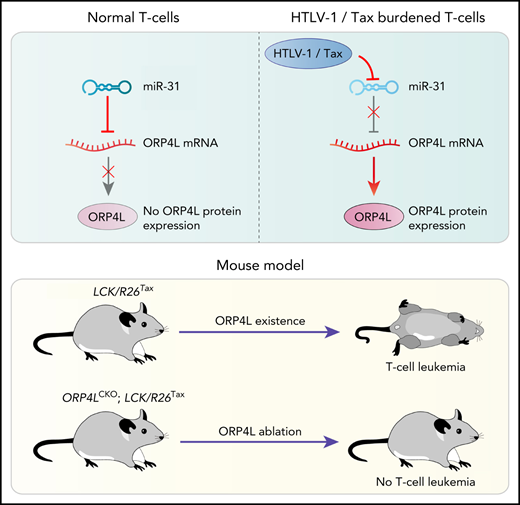
Key Points
ORP4L deletion blocks Tax-induced T-cell leukemia, whereas engineering ORP4L expression in T cells results in T-cell leukemia in mice.
Loss of miR-31 induced by Tax releases ORP4L expression, which initiates T-cell deterioration, but ORP4L inhibition eliminates ATL in PDX mice.

Abstract
Human T-cell leukemia virus 1 (HTLV-1) causes adult T-cell leukemia (ATL), but the mechanism underlying its initiation remains elusive. In this study, ORP4L was expressed in ATL cells but not in normal T-cells. ORP4L ablation completely blocked T-cell leukemogenesis induced by the HTLV-1 oncoprotein Tax in mice, whereas engineering ORP4L expression in T-cells resulted in T-cell leukemia in mice, suggesting the oncogenic properties and prerequisite of ORP4L promote the initiation of T-cell leukemogenesis. For molecular insight, we found that loss of miR-31 caused by HTLV-1 induced ORP4L expression in T-cells. ORP4L interacts with PI3Kδ to promote PI(3,4,5)P3 generation, contributing to AKT hyperactivation; NF-κB–dependent, p53 inactivation-induced pro-oncogene expression; and T-cell leukemogenesis. Consistently, ORP4L ablation eliminates human ATL cells in patient-derived xenograft ATL models. These results reveal a plausible mechanism of T-cell deterioration by HTLV-1 that can be therapeutically targeted.
Introduction
There are an estimated 5 to 20 million individuals infected with human T-cell leukemia virus 1 (HTLV-1) worldwide, and 3% to 5% of these carriers progress to deadly adult T-cell leukemia (ATL), with a median survival time of 2 to 6 months.1-3 HTLV-1 directly infects peripheral T cells, and its oncoprotein Tax induces the malignant transformation of these cells.4,5 Tax induces PI3K-AKT signaling activation and the downstream NF-κB–dependent p53 inhibition,6,7 enabling cells to acquire oncogenic properties that promote malignant transformation.8,9 However, the initial mechanisms of HTLV-1–induced carcinogenesis remain elusive.
Oxysterol-binding protein (OSBP)–related proteins (ORPs) have emerged as key mediators of nonvesicular lipid transport10,11 and metabolism of phosphoinositides.12,13 Among ORPs, ORP4 is expressed constitutively in the brain, heart, and testis, but is virtually absent from other human and murine tissues. ORP4-knockout mice exhibit teratozoospermia caused by the death of developing spermatozoa.14 Earlier studies also suggested that ORP4L can be aberrantly induced in distinct malignant cell types15-17 and is a target of the natural antiproliferative steroidal saponin OSW-1,18 suggesting the involvement of ORP4L in controlling oncogenic cell growth. Moreover, our recent works indicated that ORP4L is associated with the survival of T-cell acute lymphoblastic leukemia (T-ALL) cells19 and leukemia stem cells (LSCs)20 by mediating cellular phosphoinositide signals. However, its functional role in leukemogenesis is undefined.
Methods
Human specimens and cell lines
The study protocol was approved by the Human Ethics Review Committee of Jinan University and was performed in accordance with the Declaration of Helsinki. All samples were collected after informed consent was obtained from the patients. The clinical information of subjects is provided in supplemental Table 1 (available on the Blood Web site). Peripheral blood mononuclear cells (PBMCs) from healthy volunteers and patients with ATL were purified by Ficoll-Hypaque gradient centrifugation. The native T cells were isolated by using Enhanced Human T-Cell Immunocolumns (Cedarlane) according to the manufacturer’s instructions and were cultured in RPMI 1640 containing 20% fetal bovine serum (FBS) and 20 U/mL recombinant human interleukin-2 (IL-2; PeproTech). For HTLV-1 infection of normal human T cells, we induced infection as previously described.21 In brief, we obtained virus from MT-4 cells resuspended in fresh medium 16 hours beforehand at 2 × 106 cells per mL. We incubated dendritic cells with HTLV-1. After culturing for 3 days, we added T cells to the dendritic cells and refed the cultures with a 1:1 ratio of the medium for dendritic cells added to the medium for T cells. We separated T cells from the dendritic cells and further cultured T cells for 16 weeks in RPMI 1640 containing 20% FBS and 20 U/mL recombinant human IL-2, during which the medium was replaced every 3 days.
MT-4 and HeLa cells were purchased from American Type Culture Collection and maintained in RPMI 1640 (For MT-4 cells) or Dulbecco’s modified Eagle’s medium (for HeLa cells) containing 10% FBS, 100 U/mL penicillin, and 100 mg/mL streptomycin at 37°C in a humidified incubator with 5% CO2. The cell lines were authenticated by Promega short-tandem repeat analysis and tested for mycoplasma contamination before use in the experiments.
Generation of LCK/R26Tax and ORP4Lcko; LCK/R26Tax mouse models
The animals were housed at the Experimental Animal Center of Jinan University. All animal procedures were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee (IACUC) at Jinan University.
To generate a conditional Tax knockin at the Rosa26 locus, a targeting vector containing the 5′ homology arm, the LCK promoter-Tax-polyA fragment, and the 3′ homology arm was obtained by using in-fusion cloning methods. Cas9 and guide (gRNA) were coinjected into fertilized eggs with this targeting vector, with the leading targeting vector inserted into intron 1 of Rosa26 located on mouse chromosome 6 in reverse orientation for mouse production. The pups were genotyped by polymerase chain reaction (PCR) followed by sequencing analysis to confirm appropriate genomic targeting. To further confirm the correct homologous recombination on chromosome 6, we performed southern blot analysis using 5′ and 3′ probes with BamHI- and BstEII-digested genomic DNA blots. The mice with heterozygotic single-copy insertion were selected for breeding to generate homozygous mice with a 2-copy insertion. The mice having both floxed ORP4L alleles generated in our previous study22 were crossed with Cre transgenic mice (LCK/Cre; Jackson Laboratory stock no.: 006889) in which Cre is driven by the LCK promoter, to obtain mice specifically deficient in T-cell ORP4L (ORP4Lcko). These ORP4Lcko mice were crossed with LCK/R26Tax mice to generate ORP4L knockout heterozygous mouse lines (ORP4L−/+;LCK/R26Tax), and the mice were then inbred to generate mouse lines with Tax expression, but with or without ORP4L expression in the T cells (LCK/R26Tax and ORP4Lcko;LCK/R26Tax).
Mouse T-cell isolation and in vitro culture
Mouse T cells were isolated from peripheral blood mononuclear cells (PBMCs) or splenocytes. Whole peripheral blood was treated with red blood cell lysis buffer to obtain the PBMCs. The spleens were digested with collagenase II (Sigma) at a concentration of 0.1 U/mL for 20 minutes at 37°C, and the suspended cells were collected every 10 minutes, followed by centrifugation for 10 minutes at 1800 rpm. After that, the red blood cells were removed by using the lysis buffer. CD4+ T cells were further isolated from PBMCs or splenocytes by using a naive CD4+ T-Cell Isolation Kit (130-104-453; Miltenyi Biotec) according to the manufacturer’s instructions. These T cells were cultured in 20 U/mL IL-2 (212-1; Peprotech) and activated with 1 μg/mL phytohemagglutinin (693839; Sigma-Aldrich) as described.21,23
IL-2 independent T-cell transformation assay
IL-2 independent T-cell transformation was assayed as previously described.21,24 In brief, mouse T cells infected with lentivirus were cultured for 16 weeks in medium containing 20 U/mL IL-2, during which the medium was replaced every 3 days. 16 weeks later, the IL-2 was removed from the medium for further culturing, and the cell death rates were quantified by using the Live/Dead Fixable Near-IR Dead Cell Stain Kit (ThermoFisher).
T-cell transplantation into B-NDG mice
B-NDG mice were purchased from Biocytogen and housed at the Experimental Animal Center of Jinan University. We harvested wild-type (WT) or LCK/R26Tax CD4+ T cells and infected LCK/R26Tax T cells with lentivirus encoding control RNA or miR-31 (Figure 2I-L). Two days later, the cells were additionally infected with ORP4L-expressing lentivirus and cultured for an additional 2 days in the presence of 20 U/mL IL-2, after which they were collected and injected IV into B-NDG mice with 1.6-Gy irradiation (3 × 106 cells per mouse in 100 μL phosphate-buffered saline [PBS]). Human patient’s ATL T cells were infected with lentivirus encoding control RNA or miR-31 (Figure 2M-N; supplemental Figure 2N). Two days later, the cells were also infected with ORP4L-expressing lentivirus and cultured for an additional 2 days, after which they were collected and injected IV into B-NDG mice with 1.6-Gy irradiation (1 × 107 cells per mouse in 100 μL PBS). We harvested murine WT CD4+ T cells and infected T cells with lentivirus encoding, with or without ORP4L (Figure 3). The cells were cultured for 16 weeks in the presence of 20 U/mL IL-2, after which they were collected and injected IV into B-NDG mice with 1.6-Gy irradiation (3 × 106 cells per mouse in 100 μL PBS).
Inducible lentiviral single guide RNA expression system
To obtain highly efficient, inducible ORP4L gene knockout in leukemia T cells, we used a dual lentiviral vector system consisting of a constitutive expression vector for the Cas9 endonuclease and a doxycycline (Dox)-inducible ORP4L sgRNA cassette and promoter-driven tetracycline (Tet) repressor–linked EGFP protein via T2A peptide. The single guide RNA (sgRNA) is driven by the H1 promoter containing a Tet operator site, allowing for tight suppression of the promoter activity in the presence of a Tet repressor, which can be efficiently relieved by the addition of Dox. The sgRNA sequences can be found in supplemental Table 4.
Animals in vivo treatments
Primary human ATL cells were infected with an inducible sgRNA system, and the cells were transplanted into B-NDG–recipient mice with 1.6-Gy irradiation (1 × 107 primary human ATL cells/mouse in 100 μL PBS) and then monitored for development of T-cell leukemia. To delete ORP4L gene, Dox was administered via food pellets (625 mg/kg) 10 days after transplantation. Pathological analyses were conducted after Dox treatment (Figure 6E-F).
We isolated primary T cells from patients with ATL and then transplanted them into B-NDG–recipient mice with 1.6-Gy irradiation (1 × 107 cells per mouse in 100 μL PBS); 2 weeks later, the mice were randomly divided into 2 groups and treated with ORP4L inhibitor LYZ-81 (5.8 mg/kg per 2 days, IV) or vehicle control for a further 2 weeks. Pathological analyses were conducted after LYZ-81 treatment (Figure 6I-K).
Result
ORP4L depletion blocks the induction of T-cell leukemogenesis by the HTLV-1 oncoprotein Tax
We previously demonstrated that ORP4L is expressed in T-ALL cells but not in normal T cells.19 In the present study, we found that ORP4L is undetectable in normal naïve and activated T cells (supplemental Figure 1A), but it is highly expressed in ATL cells, regardless of the expression of Tax (Figure 1A-B). We infected human T cells by HTLV-1 or transduced them with lentivirus carrying the HTLV-1 oncogenes Tax or HBZ. ORP4L expression was dramatically induced in HTLV-1–infected or oncogene Tax-expressing human T cells, whereas HBZ did not induce ORP4L expression (Figure 1C). We next infected human T cells with lentivirus constructed with specific short hairpin RNA targeting Tax, followed by infection with HTLV-1. After 16 weeks of culturing in vitro, we analyzed the ORP4L expression and found that ORP4L expression was significantly suppressed in HTLV-1–infected T cells subjected to Tax knockdown (supplemental Figure 1B), supporting the notion that HTLV-1 induces ORP4L expression directly through Tax.
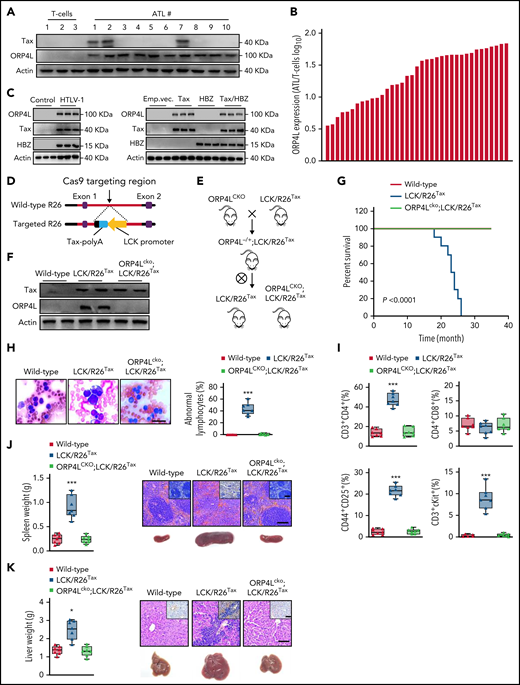
ORP4L knockout prevents HTLV-1 oncogene Tax-induced T-cell leukemia in mice. (A) Western blot analysis of Tax and ORP4L expression in normal human T cells and T cells of patients with ATL. (B) Quantitative PCR (qPCR) analysis of ORP4L expression in normal human T cells (n = 5) and human ATL T cells (n = 37). (C) Western blot analysis of ORP4L expression in HTLV-1–infected human T cells (left) and human T cells infected with a lentivirus carrying Tax, HBZ, or both. T cells were infected with HTLV-1 or transduced with lentivirus and cultured in vitro for 16 weeks. (D) The targeting construction used to generate the T-cell–expressing Tax knockin mice. (E) Schematic representation of mouse line crossing strategy. (F) Western blot analysis of the expression of ORP4L in T-cells from 2-month-old WT, LCK/R26Tax, and ORP4Lcko;LCK/R26Tax mice. (G) Kaplan-Meier comparative survival analysis of WT, LCK/R26Tax, and ORP4Lcko;LCK/R26Tax mice (n = 12 mice of each group, log-rank test). (H) Representative images of peripheral blood smears from sick LCK/R26Tax mice and WT or ORP4Lcko;LCK/R26Tax littermate mice. Morphologically abnormal leukemic cells with large nuclei were present. The number of abnormal lymphocytes on the smears from each cohort are shown. Scale bars, 100 μm. (I) The percentage of CD3+CD4+, CD4+CD8+, CD44+CD25+, and CD3+c-kit+ cells in the peripheral blood of sick LCK/R26Tax mice and WT or ORP4Lcko;LCK/R26Tax littermate mice. Mean ± standard deviation (SD; n = 7 mice of each group; Student t test). (J-K) Splenomegaly (J) and hepatomegaly (K) in sick LCK/R26Tax mice and WT or ORP4Lcko;LCK/R26Tax littermate mice. Representative histologic hematoxylin and hematoxylin eosin–stained sections show T-cell infiltration and immunohistochemical images with positive CD3-specific antibody staining. Scale bars, 100 μm. Mean ± SD (n = 7 mice of each group; Student t test). *P < .05; ***P < .001.
ORP4L knockout prevents HTLV-1 oncogene Tax-induced T-cell leukemia in mice. (A) Western blot analysis of Tax and ORP4L expression in normal human T cells and T cells of patients with ATL. (B) Quantitative PCR (qPCR) analysis of ORP4L expression in normal human T cells (n = 5) and human ATL T cells (n = 37). (C) Western blot analysis of ORP4L expression in HTLV-1–infected human T cells (left) and human T cells infected with a lentivirus carrying Tax, HBZ, or both. T cells were infected with HTLV-1 or transduced with lentivirus and cultured in vitro for 16 weeks. (D) The targeting construction used to generate the T-cell–expressing Tax knockin mice. (E) Schematic representation of mouse line crossing strategy. (F) Western blot analysis of the expression of ORP4L in T-cells from 2-month-old WT, LCK/R26Tax, and ORP4Lcko;LCK/R26Tax mice. (G) Kaplan-Meier comparative survival analysis of WT, LCK/R26Tax, and ORP4Lcko;LCK/R26Tax mice (n = 12 mice of each group, log-rank test). (H) Representative images of peripheral blood smears from sick LCK/R26Tax mice and WT or ORP4Lcko;LCK/R26Tax littermate mice. Morphologically abnormal leukemic cells with large nuclei were present. The number of abnormal lymphocytes on the smears from each cohort are shown. Scale bars, 100 μm. (I) The percentage of CD3+CD4+, CD4+CD8+, CD44+CD25+, and CD3+c-kit+ cells in the peripheral blood of sick LCK/R26Tax mice and WT or ORP4Lcko;LCK/R26Tax littermate mice. Mean ± standard deviation (SD; n = 7 mice of each group; Student t test). (J-K) Splenomegaly (J) and hepatomegaly (K) in sick LCK/R26Tax mice and WT or ORP4Lcko;LCK/R26Tax littermate mice. Representative histologic hematoxylin and hematoxylin eosin–stained sections show T-cell infiltration and immunohistochemical images with positive CD3-specific antibody staining. Scale bars, 100 μm. Mean ± SD (n = 7 mice of each group; Student t test). *P < .05; ***P < .001.
To evaluate the role of ORP4L in T-cell leukemogenesis, we generated, by CRISPR/Cas9 genome editing in the Rosa26 locus, a knockin mouse expressing Tax under the control of the LCK proximal promoter, which restricts Tax expression in developing thymocytes (LCK/R26Tax mice; Figure 1D). We confirmed the correct Tax homologous recombination by Southern blot (supplemental Figure 1C-D). Mice with both floxed ORP4L alleles were generated in our previous study22 and were crossed with LCK/Cre transgenic mice to obtain animals with ORP4L-specific deficiency in T-cells (ORP4Lcko). These ORP4Lcko mice have a normal lifespan, no disease, and normal peripheral blood cell counts, including CD3+ T cells, CD19+ B cells, and Gr-1+ granulocyte cells (supplemental Figure 1E). The LCK/R26Tax mice were further crossed with ORP4Lcko to generate ORP4Lcko;LCK/R26Tax mice with Tax expression and ORP4L deletion in T-cells (Figure 1E). ORP4L expression was detected in T cells from the LCK/R26Tax mice at the age of 2, 6, and 12 months (Figure 1F; supplemental Figure 1F). These results demonstrated that HTLV-one with Tax induces ORP4L expression in T-cells. Tax expression is frequently lost from ATLs by genetic changes or epigenetic modifications of the proviral genome.25 To evaluate whether ORP4L expression may cause Tax silencing, we knocked down ORP4L in T cells from LCK/R26Tax mice and primary T cells from patients with ATL with Tax expression. ORP4L depletion did not affect the expression of Tax in T cells from LCK/R26Tax mice (supplemental Figure 1G) and patients with ATL (supplemental Figure 1H), suggesting that Tax expression does not depend on ORP4L. In contrast, we knocked down Tax expression in T cells from LCK/R26Tax mice at the age of 6 months and found that it did not change ORP4L expression (supplemental Figure 1I).
Tax transgenic mice have been reported to develop T-cell leukemia.26-28 LCK/R26Tax mice developed T-cell leukemia at the age of ∼18 months and died within 25 months, whereas ORP4Lcko;LCK/R26Tax mice maintained a healthy status (Figure 1G). We also observed leukemic cells with a cleaved nucleus morphology in LCK/R26Tax mice, morphologically identical to the “flower” cells in human ATL, albeit at low incidence (<2%; supplemental Figure 1J). ORP4L ablation completely blocked Tax-induced T-cell leukemogenesis phenotypes, including the increasing number of large and abnormal leukemic cells in peripheral blood (Figure 1H); the increase of CD4+, CD44+CD25+, and c-Kit+ T cells (Figure 1I); and the splenomegaly (Figure 1J) and hepatomegaly (Figure 1K) with leukemic T-cell infiltration. These results strongly indicate that ORP4L is a prerequisite for Tax-induced T-cell leukemia in mice.
Tax-induced loss of miR-31 results in ORP4L expression and T-cell leukemia
We next investigated how HTLV-1/Tax induces ORP4L expression. TargetScan prediction indicated that both the human and murine ORP4L 3′UTR are directly targeted by miR-31 (supplemental Figure 2A-B). ORP4L 3′UTR luciferase reporter (supplemental Figure 2C) assays revealed that miR-31 mimic decreased the reporter activity, whereas miR-31 inhibitor increased it (supplemental Figure 2D). In contrast, a reporter with the miR-31 seed region mutated was not affected by miR-31 (supplemental Figure 2D). Consistently, miR-31 inhibitor elevated the expression of ORP4L in mRNA (Figure 2A) and protein (Figure 2B), whereas the miR-31 mimic reduced ORP4 expression. Because miR-31 was genetically lost in ATL cells,29 we hypothesized that Tax may induce ORP4L expression via suppression of miR-31. Indeed, miR-31 expression was dramatically reduced in the T-cells of a patient with ATL (Figure 2C). Pearson’s correlation coefficient within ATL cells revealed that ORP4L mRNA levels correlated negatively with miR-31 (Figure 2D). miR-31 was reduced by HTLV-1 infection or Tax expression in normal human T-cells (supplemental Figure 2E). To examine whether miR-31 inhibits ORP4L expression through the seed sequence, we constructed plasmid ORP4L expression vectors without 3′UTR (Flag-ORP4L), with 3′UTR (Flag-ORP4L-WT3′UTR), and with mutated 3′UTR (Flag-ORP4L-Mu3′UTR) and tested their expressions in HeLa cells. Results showed that expression of Flag-ORP4L-WT3′UTR was suppressed by simultaneous introduction of miR-31 mimic, whereas miR-31 inhibition inversely upregulated the ORP4L level. In contrast, these observations were absent in Flag-ORP4L-Mu3′UTR transfected cells, revealing that the cellular miR-31 level negatively affected ORP4L expression through its 3′UTR sequence (supplemental Figure 2F). Indeed, a multicolor fluorescence in situ hybridization assay of samples from 3 mice and genomic region amplification by PCR of samples from 15 mice showed that the miR-31–encoding genomic region was lost in T-cells, but not in B cells or macrophages from LCK/R26Tax mice (Figure 2E; supplemental Figure 2G-H), accompanied by dramatically reduced miR-31 expression in LCK/R26Tax T-cells (Figure 2F). In addition, ectopic miR-31 expression blocked ORP4L expression in LCK/R26Tax T cells (Figure 2G; supplemental Figure 2I) and in T-cells of patient ATL#21 (Figure 2H; supplemental Figure 2J-M), indicating that suppression of miR-31 is the major event underlying Tax-induced ORP4L expression.
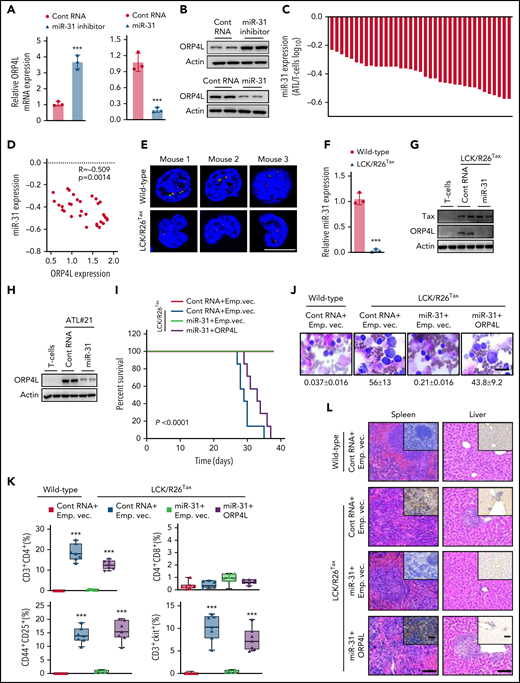
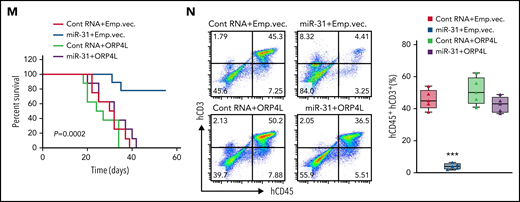
Tax triggers ORP4L expression and T-cell malignant transformation via suppression of miR-31. (A-B) qPCR (A) and western blot (B) analyses confirming the effects of miR-31 inhibitor on ORP4L expression in HeLa cells and the effects of miR-31 mimic on ORP4L expression in MT-4 cells. Mean ± standard deviation (SD; n = 3 experimental repeat; Student t test). (C) qPCR analysis of miR-31 expression in T cells of patients with ATL (n = 37), as compared with normal T cells (n = 5). (D) Pearson’s correlation coefficient of miR-31 and ORP4L expression within human ATL specimens (n = 37). (E) Multicolor fluorescence in situ hybridization analysis of the genomic status of miR-31 encoding (green probe) and its host gene (origin probe) regions in T cells of 2-month-old WT and LCK/R26Tax mice. The chromosome 4 centromere (red probe) is included as a control. Scale bar, 5 μm. (F) miR-31 expression in 2-month-old WT and LCK/R26Tax mouse T cells analyzed by qPCR. Mean ± SD (n = 3 mice of each group; Student t test). (G) Western blot analysis of the expression of ORP4L in LCK/R26Tax T cells or LCK/R26Tax T cells overexpressing miR-31. The cells were isolated and infected with lentivirus carrying control RNA or miR-31 and cultured for 96 hours in vitro. (H) Western blot analysis of the expression of ORP4L in human normal T cells or human ATL T cells overexpressing miR-31. The cells were isolated and infected with lentivirus carrying control RNA or miR-31 and cultured for 96 hours in vitro. (I) Kaplan-Meier comparative survival analysis of B-NDG mouse recipients of WT T cells or LCK/R26Tax T cells overexpressing miR-31 or overexpressing both miR-31 and ORP4L. The LCK/R26Tax T cells were coinfected with the lentivirus carrying control RNA miR-31, empty vector (Emp.vec.), or ORP4L as follows: control RNA+Emp.vec., miR-31+Emp.vec., miR-31+ORP4L, and cultured for 96 hours in vitro before transplantation (n = 7; mice of each group, log-rank test). (J) Representative images of peripheral blood smears from B-NDG mice treated as in panel I. The number of abnormal lymphocytes on smears in each cohort are indicated below the images. Scale bar, 10 μm. (K) The percentage of CD3+CD4+, CD4+CD8+, CD44+CD25+, and CD3+c-Kit+ cells in peripheral blood of B-NDG mice treated as in panel I. Mean ± SD (n = 7 mice of each group; Student t test). (L) Representative histologic hematoxylin and eosin– and anti-CD3–stained sections showing T-cell infiltration in spleen and liver of B-NDG mice treated as in panel I. Scale bars, 100 μm. (M) Kaplan-Meier comparative survival analysis of B-NDG mouse recipients of T cells of patients with ATL (ATL#21). The cells were isolated and coinfected with lentivirus carrying control RNA, miR-31, Emp.vec. or ORP4L as follows: control RNA+Emp.vec., miR-31+Emp.vec., control RNA+ORP4L, and miR-31+ORP4L and cultured for 96 hours in vitro before transplantation (n = 8 mice each group, log-rank test). (N) The percentage of human CD45+CD3+ ATL T cells in peripheral blood of B-NDG mice treated as in panel M. Mean ± SD (n = 8 mice in each group; Student t test). ***P < .001.
Tax triggers ORP4L expression and T-cell malignant transformation via suppression of miR-31. (A-B) qPCR (A) and western blot (B) analyses confirming the effects of miR-31 inhibitor on ORP4L expression in HeLa cells and the effects of miR-31 mimic on ORP4L expression in MT-4 cells. Mean ± standard deviation (SD; n = 3 experimental repeat; Student t test). (C) qPCR analysis of miR-31 expression in T cells of patients with ATL (n = 37), as compared with normal T cells (n = 5). (D) Pearson’s correlation coefficient of miR-31 and ORP4L expression within human ATL specimens (n = 37). (E) Multicolor fluorescence in situ hybridization analysis of the genomic status of miR-31 encoding (green probe) and its host gene (origin probe) regions in T cells of 2-month-old WT and LCK/R26Tax mice. The chromosome 4 centromere (red probe) is included as a control. Scale bar, 5 μm. (F) miR-31 expression in 2-month-old WT and LCK/R26Tax mouse T cells analyzed by qPCR. Mean ± SD (n = 3 mice of each group; Student t test). (G) Western blot analysis of the expression of ORP4L in LCK/R26Tax T cells or LCK/R26Tax T cells overexpressing miR-31. The cells were isolated and infected with lentivirus carrying control RNA or miR-31 and cultured for 96 hours in vitro. (H) Western blot analysis of the expression of ORP4L in human normal T cells or human ATL T cells overexpressing miR-31. The cells were isolated and infected with lentivirus carrying control RNA or miR-31 and cultured for 96 hours in vitro. (I) Kaplan-Meier comparative survival analysis of B-NDG mouse recipients of WT T cells or LCK/R26Tax T cells overexpressing miR-31 or overexpressing both miR-31 and ORP4L. The LCK/R26Tax T cells were coinfected with the lentivirus carrying control RNA miR-31, empty vector (Emp.vec.), or ORP4L as follows: control RNA+Emp.vec., miR-31+Emp.vec., miR-31+ORP4L, and cultured for 96 hours in vitro before transplantation (n = 7; mice of each group, log-rank test). (J) Representative images of peripheral blood smears from B-NDG mice treated as in panel I. The number of abnormal lymphocytes on smears in each cohort are indicated below the images. Scale bar, 10 μm. (K) The percentage of CD3+CD4+, CD4+CD8+, CD44+CD25+, and CD3+c-Kit+ cells in peripheral blood of B-NDG mice treated as in panel I. Mean ± SD (n = 7 mice of each group; Student t test). (L) Representative histologic hematoxylin and eosin– and anti-CD3–stained sections showing T-cell infiltration in spleen and liver of B-NDG mice treated as in panel I. Scale bars, 100 μm. (M) Kaplan-Meier comparative survival analysis of B-NDG mouse recipients of T cells of patients with ATL (ATL#21). The cells were isolated and coinfected with lentivirus carrying control RNA, miR-31, Emp.vec. or ORP4L as follows: control RNA+Emp.vec., miR-31+Emp.vec., control RNA+ORP4L, and miR-31+ORP4L and cultured for 96 hours in vitro before transplantation (n = 8 mice each group, log-rank test). (N) The percentage of human CD45+CD3+ ATL T cells in peripheral blood of B-NDG mice treated as in panel M. Mean ± SD (n = 8 mice in each group; Student t test). ***P < .001.
To verify the role of the miR-31 loss/ORP4L gain axis in Tax-induced T-cell leukemia, we infected LCK/R26Tax T cells from 10-month-old mice with lentivirus encoding miR-31 or ORP4L and then transplanted them into B-NDG mice to test leukemogenesis. LCK/R26Tax T-cell transplantation resulted in T-cell leukemia and death of the mice within 5 weeks, which were prevented by ectopic miR-31 expression, but were reelicited by ORP4L expression (Figure 2I-L). Similarly, we infected T cells of patients with ATL with lentivirus encoding miR-31 or ORP4L and then transplanted them into B-NDG mice. miR-31 expression prolonged animal survival (Figure 2M; supplemental Figure 2N-O) and reduced ATL cell engraftment (Figure 2N), whereas the protective effect was abolished by expression of ORP4L. These observations demonstrate that suppression of miR-31 results in the expression of ORP4L, driving Tax-induced T-cell leukemia transformation.
Engineering ORP4L expression induces T-cell malignant transformation and T-cell leukemia in mice
To further verify the role of ORP4L in T-cell leukemia, we transfected normal murine T cells with lentivirus encoding ORP4L and cultured them in vitro (Figure 3A). Transition from IL-2–dependent to –independent growth is a key step in the transformation of HTLV-1–infected T cells.30 After culturing for 16 weeks, T cells with ORP4L expression continued to proliferate and survive after the removal of IL-2 from the medium, whereas T cells without ORP4L expression rapidly died (Figure 3B). We transplanted these T cells into B-NDG mice to test development of leukemia. Transplantation of ORP4L-expressing T cells resulted in the death of the mice within 9 weeks (Figure 3C). We next characterized the major phenotype of these morbid B-NDG mice and revealed the increased CD3+CD4+ T cells in PBMCs. Further analysis showed that these cells included CD44+CD25+ and c-Kit+ T cells (Figure 3D). Peripheral blood smears from these mice showed the presence of large and abnormal leukemic cells in a moribund state (Figure 3E). The mouse recipients of ORP4L-expressing T cells presented with the development of marked splenomegaly (Figure 3F) and hepatomegaly (Figure 3G). Histological analysis revealed diffuse, large-cell lymphomatous infiltration in the spleen, liver, and lung (Figure 3H). Immunohistochemical staining identified these infiltrated cells as CD3+ T cells (Figure 3H). We tested and found that ORP4L was expressed in lentivirus-infected T cells before transplantation (Figure 3I; top) and T cells isolated from morbid B-NDG mice after transplantation (Figure 3I; bottom), indicating the sustained expression of ORP4L in vivo in this experiment. The pathology shown by the mouse recipients of ORP4L-expressing T cells represents T-cell leukemia similar to Tax-induced leukemic symptoms, thus providing evidence for the leukemogenic potential of ORP4L.
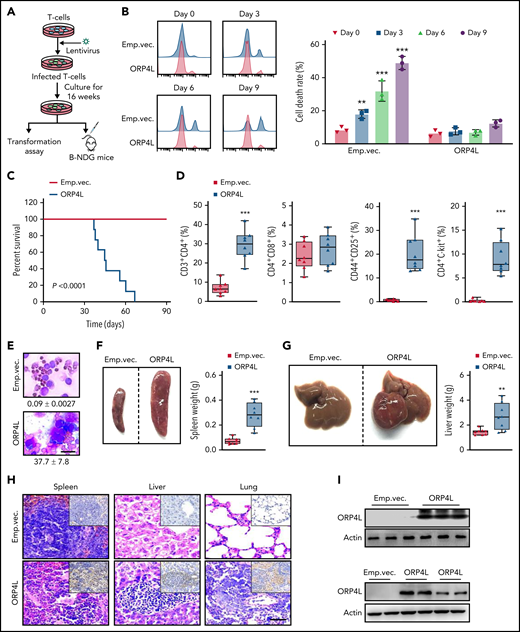
ORP4L expression induces T-cell malignant transformation and T-cell leukemogenesis in mice. (A) The experimental design. Normal murine CD4+ T cells were infected with lentivirus with or without ORP4L expression and cultured for 16 weeks in vitro; then, the cells were used for malignant transformation assay or B-NDG mice transplantation. (B) Cell death analysis of T cells infected with lentivirus carrying ORP4L or not. After culture for 16 weeks in vitro, the cell death rates were detected at 0, 3, 6, and 9 days after removing the IL-2 from the culture medium. Mean ± standard deviation (SD; n = 3 experiments; Student t test). (C) Kaplan-Meier comparative survival analysis of B-NDG mouse recipients of T-cell transplants with or without ORP4L expression (n = 8 mice each group, log-rank test). (D) The percentage of CD3+CD4+, CD4+CD8+, CD44+CD25+, and CD3+c-Kit+ cells in the peripheral blood of B-NDG mice treated as in panel C (n = 8 mice each group; Student t test). (E) Representative images of peripheral blood smear from B-NDG mice treated as in panel C. The number of abnormal lymphocytes is indicated below the smears for both cohorts. Bar represents 10 μm. (F-G) Splenomegaly (F) and hepatomegaly (G) in B-NDG mice treated as in panel C. Representative organs are shown on the left, with organ weights on the right. Mean ± SD (n = 8 mice each group; Student t test). (H) Representative histologic hematoxylin and eosin–stained sections showing T-cell infiltration in spleen, liver, and lung of B-NDG mice treated as in panel C. Immunohistochemical human CD3-specific antibody staining (insets) of the tissues are shown. Bar represents 100 μm. (I) Western blot analysis of ORP4L expression in T cells. ORP4L expression in T cells infected with lentivirus with or without ORP4L before transplantation (top) or ORP4L expression in T cells isolated from 2 B-NDG mice after transplantation when the mice are sick (bottom). **P < .01; ***P < .001.
ORP4L expression induces T-cell malignant transformation and T-cell leukemogenesis in mice. (A) The experimental design. Normal murine CD4+ T cells were infected with lentivirus with or without ORP4L expression and cultured for 16 weeks in vitro; then, the cells were used for malignant transformation assay or B-NDG mice transplantation. (B) Cell death analysis of T cells infected with lentivirus carrying ORP4L or not. After culture for 16 weeks in vitro, the cell death rates were detected at 0, 3, 6, and 9 days after removing the IL-2 from the culture medium. Mean ± standard deviation (SD; n = 3 experiments; Student t test). (C) Kaplan-Meier comparative survival analysis of B-NDG mouse recipients of T-cell transplants with or without ORP4L expression (n = 8 mice each group, log-rank test). (D) The percentage of CD3+CD4+, CD4+CD8+, CD44+CD25+, and CD3+c-Kit+ cells in the peripheral blood of B-NDG mice treated as in panel C (n = 8 mice each group; Student t test). (E) Representative images of peripheral blood smear from B-NDG mice treated as in panel C. The number of abnormal lymphocytes is indicated below the smears for both cohorts. Bar represents 10 μm. (F-G) Splenomegaly (F) and hepatomegaly (G) in B-NDG mice treated as in panel C. Representative organs are shown on the left, with organ weights on the right. Mean ± SD (n = 8 mice each group; Student t test). (H) Representative histologic hematoxylin and eosin–stained sections showing T-cell infiltration in spleen, liver, and lung of B-NDG mice treated as in panel C. Immunohistochemical human CD3-specific antibody staining (insets) of the tissues are shown. Bar represents 100 μm. (I) Western blot analysis of ORP4L expression in T cells. ORP4L expression in T cells infected with lentivirus with or without ORP4L before transplantation (top) or ORP4L expression in T cells isolated from 2 B-NDG mice after transplantation when the mice are sick (bottom). **P < .01; ***P < .001.
ORP4L interacts with PI3Kδ to promote PI(3,4,5)P3 generation and AKT activation
ORPs are essential mediators in the intracellular transport and metabolism of phosphoinositides.12,13 Thus, we focused on the role of plasma membrane (PM) phosphoinositides regulated by ORP4L. Yeast 2-hybrid screening identified PI3Kδ, a PI3K isoform in leukocytes31 that is dysregulated in T-cell leukemia,32 as an interaction partner of ORP4L (supplemental Figure 3A). Coimmunoprecipitation and immunofluorescence microscopy further verified the interaction and colocalization of these 2 proteins in MT-4 cells (Figure 4A). The amino acid residues 422 to 495 in ORP4L were required for binding to PI3Kδ, as determined by the yeast 2-hybrid assay (supplemental Figure 3A). Prediction models of ORP4L (aa 422-495) and PI3Kδ (PDB: 6G6W) suggested that ORP4L interacts with PI3Kδ through 4 ionic and 6 H-bonds (Figure 4B; supplemental Figure 3B). In supporting this possibility, ORP4L with mutations of the 4 residues at the ionic bonding sites (ORP4L△PI3Kδ) failed to bind PI3Kδ (supplemental Figure 3C).
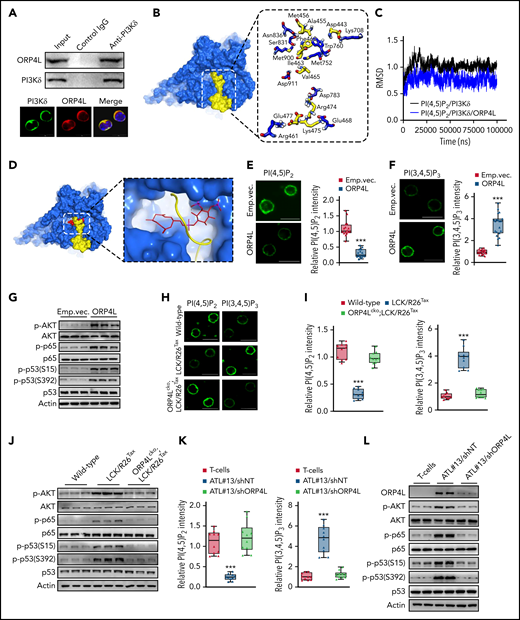
ORP4L interacts with PI3Kδ to enhance PI(3,4,5)P3 generation and AKT activation in malignant transformed T cells. (A) Coimmunoprecipitation (top) and confocal microscopy (bottom) analysis of ORP4L binding to and colocalization with PI3Kδ in MT-4 cells. Scale bar, 10 μm. (B) The binding model of ORP4L (yellow) to PI3Kδ (blue) computationally predicted. The key interaction residues of ORP4L (yellow) and PI3Kδ (blue) complex is shown. (C) The root-mean-square deviation values of PI(4,5)P2 in PI3Kδ and in the ORP4L-PI3Kδ complex. (D) The binding model of ORP4L (yellow) to PI(4,5)P2 (red)/PI3Kδ (blue) complex computationally predicted. The site of PI(4,5)P2/PI3Kδ/ORP4L complex is shown. (E-F) PI(4,5)P2 (E) and PI(3,4,5)P3 (F) contents in T cells isolated from sick B-NDG mice in Figure 3. Scale bars, 10 μm. The panels on the right indicate quantitation of relative fluorescence intensity. Mean ± standard deviation (SD; n = 15-20 cells; Student t test). (G) Phosphorylated AKT, p65, and p53 levels in T cells isolated from the sick B-NDG mice in Figure 3. (H-I) PI(4,5)P2 and PI(3,4,5)P3 contents in T cells of WT, LCK/R26Tax, and ORP4Lcko;LCK/R26Tax mice at the age of 15 months. Representative images (H) and the relative quantification of fluorescence intensity (I) are shown. Scale bars, 10 μm. Mean ± SD (n = 10 cells; Student t test). (J) AKT activation and p65 and p53 phosphorylation in T cells of WT, LCK/R26Tax, and ORP4Lcko;LCK/R26Tax 15-month-old mice. (K) PI(4,5)P2 and PI(3,4,5)P3 contents in human normal T cells or the T cells of a patient with ATL (ATL#13) transduced with lentivirus-carrying control (shNT) or ORP4L-specific short hairpin RNA (shORP4L). The T cells were transduced and cultured in vitro for 72 hours before analysis. Mean ± SD (n = 10 cells; Student t test). (L) AKT activation, p65 and p53 phosphorylation in T cells treated as in panel K. ***P < .001.
ORP4L interacts with PI3Kδ to enhance PI(3,4,5)P3 generation and AKT activation in malignant transformed T cells. (A) Coimmunoprecipitation (top) and confocal microscopy (bottom) analysis of ORP4L binding to and colocalization with PI3Kδ in MT-4 cells. Scale bar, 10 μm. (B) The binding model of ORP4L (yellow) to PI3Kδ (blue) computationally predicted. The key interaction residues of ORP4L (yellow) and PI3Kδ (blue) complex is shown. (C) The root-mean-square deviation values of PI(4,5)P2 in PI3Kδ and in the ORP4L-PI3Kδ complex. (D) The binding model of ORP4L (yellow) to PI(4,5)P2 (red)/PI3Kδ (blue) complex computationally predicted. The site of PI(4,5)P2/PI3Kδ/ORP4L complex is shown. (E-F) PI(4,5)P2 (E) and PI(3,4,5)P3 (F) contents in T cells isolated from sick B-NDG mice in Figure 3. Scale bars, 10 μm. The panels on the right indicate quantitation of relative fluorescence intensity. Mean ± standard deviation (SD; n = 15-20 cells; Student t test). (G) Phosphorylated AKT, p65, and p53 levels in T cells isolated from the sick B-NDG mice in Figure 3. (H-I) PI(4,5)P2 and PI(3,4,5)P3 contents in T cells of WT, LCK/R26Tax, and ORP4Lcko;LCK/R26Tax mice at the age of 15 months. Representative images (H) and the relative quantification of fluorescence intensity (I) are shown. Scale bars, 10 μm. Mean ± SD (n = 10 cells; Student t test). (J) AKT activation and p65 and p53 phosphorylation in T cells of WT, LCK/R26Tax, and ORP4Lcko;LCK/R26Tax 15-month-old mice. (K) PI(4,5)P2 and PI(3,4,5)P3 contents in human normal T cells or the T cells of a patient with ATL (ATL#13) transduced with lentivirus-carrying control (shNT) or ORP4L-specific short hairpin RNA (shORP4L). The T cells were transduced and cultured in vitro for 72 hours before analysis. Mean ± SD (n = 10 cells; Student t test). (L) AKT activation, p65 and p53 phosphorylation in T cells treated as in panel K. ***P < .001.
PI3Kδ catalyzes the production of PI(3,4,5)P3 from PI(4,5)P2. Root-mean-square deviation trajectories (Figure 4C) and the free energy of binding ΔGbind,pred (supplemental Figure 3D) indicated that PI(4,5)P2 binds more favorably to the PI3Kδ-ORP4L complex than to PI3Kδ alone. The bound PI(4,5)P2 was found to be covered by ORP4L in the modeled PI3Kδ-ORP4L complex (Figure 4D). We then detected the catalytic substrate and product of PI3Kδ in PM. PI(4,5)P2 content was reduced, whereas PI(3,4,5)P3 content was significantly elevated in ORP4L-expressing T cells isolated from B-NDG mice (Figure 4E-F). PI3Kδ overexpression increased the PM PI(3,4,5)P3 content, which was abolished in ORP4L knockdown cells (supplemental Figure 3E). Similarly, ORP4L overexpression increased PI(3,4,5)P3, whereas ORP4L△PI3Kδ failed to stimulate the level of PI(3,4,5)P3 (supplemental Figure 3F). These data strongly suggest that ORP4L is essential for PI(4,5)P2 binding to PI3Kδ and its conversion to PI(3,4,5)P3.
Human ATL is associated with elevated PI3K/AKT activity, resulting in oncogenic properties through NF-κB–dependent p53 inhibition (phosphorylated at S15 and S392).6,7 Consistent with the increased PI(3,4,5)P3 production, ORP4L-expressing T cells displayed elevated AKT and NF-κB activation, as well as inhibition of p53 (Figure 4G). Similar results of PI(4,5)P2 and PI(3,4,5)P3 contents (Figure 4H-I) and AKT/NF-κB/p53 signaling (Figure 4J) were observed in T cells of LCK/R26Tax mice, but were abolished in T cells of ORP4Lcko;LCK/R26Tax mice. Furthermore, a decrease in PI(4,5)P2 and an increase in PI(3,4,5)P3 (Figure 4K), as well as accelerated AKT/NF-κB/p53 pathway activation (Figure 4L) dependent on ORP4L expression, were detected in the T cells of patients with ATL. To stress the link between ORP4L and AKT/NF-κB/p53 signaling, we used the AKT inhibitor LY294002 and the NF-κB inhibitor TPCA-1 to treat T cells isolated from LCK/R26Tax, ORP4L expressing B-NDG mice and patients with ATL. AKT and p65 inhibition both resulted in suppression of p65 and p53 phosphorylation, as well as the expression of their target genes (supplemental Figure 4A-D). In addition, TCR engagement by anti-CD3 stimulation in human ATL T cells triggered activation of AKT/NF-κB/p53 signaling, but significantly inhibited by ORP4L knockdown (supplemental Figure 4E). These results revealed that enforced ORP4L expression in ATL T cells leads to the alteration of PM phosphoinositide homeostasis and the subsequent AKT stimulation, NF-κB activation, and p53 inhibition.
Identification of differentially expressed genes in LCK/R26Tax/ORP4L-expressing cells, and T cells of patients with ATL
To determine whether specific gene sets are dysregulated in the LCK/R26Tax/ORP4L-expressing T cells, we performed whole-transcriptome RNA sequencing. The transcriptome reprogramming was visualized by clustering analysis in ORP4L-expressing and LCK/R26Tax T cells (Figure 5A-B). Notably, deletion of ORP4L “normalized” most of the reprogrammed gene signatures induced by Tax (Figure 5B). To establish the relevance of these 2 mouse models of human ATL, we collected raw data from the microarray analysis of 19 patients with ATL by Pise-Masison et al.33 Subsequently, the Venn diagram software was used to identify DEGs commonly present in all the 3 data sets, revealing a total of 51 commonly deregulated DEGs (Figure 5C). KOBAS (KEGG Orthology-Based Annotation System) analysis revealed that these DEGs were mainly enriched in the cell cycle, pyrimidine metabolism, and p53 signaling pathways (Figure 5D). The 51 DEGs were further analyzed by using the STRING database to explore the protein-protein interaction (PPI) networks, with a special focus on hub genes that play key roles in the formation of T-cell leukemias. A network with a total of 28 nodes and 154 interactions of the overlapping DEGs was constructed (Figure 5E), and Cytoscape was used to obtain the most significant module (Figure 5F). Considering the criteria role of the AKT/NF-κB/p53 pathway in ATL, 3 p53 downstream target genes, CCNB1, CCNB2, and RRM2,34 were identified as hub genes in the PPI network (Figure 5F). We further analyzed the data of 19 patients with ATL in Pise-Masison et al33 and found that these 3 genes were upregulated in T cells of patients with ATL (Figure 5G). In line with this, expression of CCNB1, CCNB2, and RRM2 were upregulated in T cells from our ATL samples (Figure 5H-I), LCK/R26Tax mice (Figure 5J), and ORP4L-expressing mice (Figure 5K), but were attenuated by ORP4L knockdown or treatment with the p53 inhibitor PFTα (Figure 5H-K). These data indicate an ORP4L-dependent p53 inhibition and gene reprogramming related to the malignant transformation of T cells.
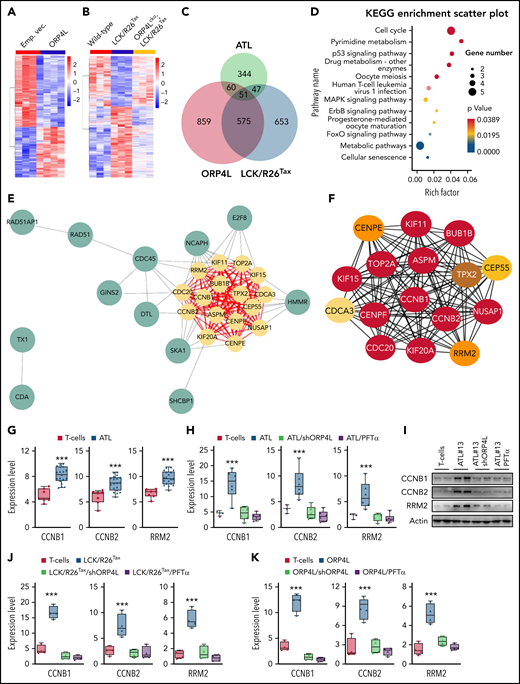
Comparison of the abnormal gene expression profiles in LCK/R26Tax- and ORP4L-expressing T cells and human ATL T cells. (A) Heat map presentation of RNA-seq data from T cells with or without ORP4L expression from sick B-NDG mice in Figure 3. (B) Heat map presentation of RNA seq data from T cells of 16-month-old WT, LCK/R26Tax, and ORP4Lcko;LCK/R26Tax mice. (C) Venn diagram showing the overlap DEGs of T cells from human ATL, T cells from LCK/R26Tax mice, and T cells with ORP4L expression from the sick B-NDG mice in Figure 3. A total of 51 overlapping genes were identified from the 3 data sets within the Venn diagrams. (D) Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis of the 51 overlapping genes in panel C. (E-F) PPI network, submodules, and hub genes. (E) The PPI network of 51 overlapping genes and clustering module (MCODE score, 15.333). Yellow circles represent hub genes. (F) Important degree of genes in the clustering modules, according to multiple consensus clustering analysis methods by the CytoHubba application. The darker the color, the more important it is. (G) Expression of p53 downstream targeting genes CCNB1, CCNB2, and RRM2 in normal human T cells and patients with ATL T cells. The data are from the literature (Pise-Masison et al33). Mean ± standard deviation (SD; n = 7 normal specimens; n = 19 specimens from patient with ATL; Student t test). (H-I) qPCR (H) and western blot (I) analyses of the expression of CCNB1, CCNB2, and RRM2 in patients with ATL (ATL 13, 16, 19, 23, 25, and 30) T cells subjected to ORP4L knockdown or p53 inhibitor PFTα. Patients with ATL T cells were transfected with ORP4L shRNA for 72 hours or treated with 50 μM PFTα for 24 hours. Mean ± SD (n = 3 normal specimens, n = 6 specimens from patient with ATL for qPCR; Student t test). (J) qPCR analysis the expression of CCNB1, CCNB2, and RRM2 in LCK/R26Tax T cells subjected to ORP4L knockdown or p53 inhibitor PFTα. T cells were transfected with ORP4L shRNA for 72 hours or treated with 50 μM PFTα for 24 hours. Mean ± SD (n = 4 mice of each group; Student t test). (K) qPCR analysis the expression of CCNB1, CCNB2, and RRM2 in T cells, with or without ORP4L expression from the sick B-NDG mice in Figure 3. T cells were transfected with ORP4L shRNA for 72 hours or treated with 50 μM PFTα for 24 hours. Mean ± SD (n = 4 mice of each group; Student t test). ***P < .001.
Comparison of the abnormal gene expression profiles in LCK/R26Tax- and ORP4L-expressing T cells and human ATL T cells. (A) Heat map presentation of RNA-seq data from T cells with or without ORP4L expression from sick B-NDG mice in Figure 3. (B) Heat map presentation of RNA seq data from T cells of 16-month-old WT, LCK/R26Tax, and ORP4Lcko;LCK/R26Tax mice. (C) Venn diagram showing the overlap DEGs of T cells from human ATL, T cells from LCK/R26Tax mice, and T cells with ORP4L expression from the sick B-NDG mice in Figure 3. A total of 51 overlapping genes were identified from the 3 data sets within the Venn diagrams. (D) Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis of the 51 overlapping genes in panel C. (E-F) PPI network, submodules, and hub genes. (E) The PPI network of 51 overlapping genes and clustering module (MCODE score, 15.333). Yellow circles represent hub genes. (F) Important degree of genes in the clustering modules, according to multiple consensus clustering analysis methods by the CytoHubba application. The darker the color, the more important it is. (G) Expression of p53 downstream targeting genes CCNB1, CCNB2, and RRM2 in normal human T cells and patients with ATL T cells. The data are from the literature (Pise-Masison et al33). Mean ± standard deviation (SD; n = 7 normal specimens; n = 19 specimens from patient with ATL; Student t test). (H-I) qPCR (H) and western blot (I) analyses of the expression of CCNB1, CCNB2, and RRM2 in patients with ATL (ATL 13, 16, 19, 23, 25, and 30) T cells subjected to ORP4L knockdown or p53 inhibitor PFTα. Patients with ATL T cells were transfected with ORP4L shRNA for 72 hours or treated with 50 μM PFTα for 24 hours. Mean ± SD (n = 3 normal specimens, n = 6 specimens from patient with ATL for qPCR; Student t test). (J) qPCR analysis the expression of CCNB1, CCNB2, and RRM2 in LCK/R26Tax T cells subjected to ORP4L knockdown or p53 inhibitor PFTα. T cells were transfected with ORP4L shRNA for 72 hours or treated with 50 μM PFTα for 24 hours. Mean ± SD (n = 4 mice of each group; Student t test). (K) qPCR analysis the expression of CCNB1, CCNB2, and RRM2 in T cells, with or without ORP4L expression from the sick B-NDG mice in Figure 3. T cells were transfected with ORP4L shRNA for 72 hours or treated with 50 μM PFTα for 24 hours. Mean ± SD (n = 4 mice of each group; Student t test). ***P < .001.
ORP4L inhibition eliminates T-cell leukemia in patient-derived xenograft ATL models
ORP4L is associated with T-ALL cell19 and LSC20 survival. We next evaluated the effect of ORP4L on ATL cell survival. We constructed a Dox-inducible CRISPR/Cas9 system to knock out ORP4L in human ATL T cells (Figure 6A). We analyzed the CD4+CD25+ subpopulation and the P (CADM1−/CD7+) subpopulation (normal T cells) progress into the N (CADM1+/CD7−) via the D (CADM1+/CD7dim+) subpopulation (supplemental Figure 5A) based on CADM1 and CD7 expression status35 in PBMCs from patients with smoldering and acute ATL, showing the phenotypes of leukemic surface markers that were compatible with the ones that are typical in ATL cells. CD4+ T cells in patients with ATL were infected with lentivirus carrying this CRISPR/Cas9 system and used for analysis in vitro and in vivo (Figure 6B). Induction of ORP4L sgRNA with Dox led to efficient reduction of the ORP4L protein and AKT/NF-κB/p53 pathway activity (Figure 6C), but to an increase in ATL cell death in vitro (Figure 6D). We then transplanted ATL cells from these patients carrying ORP4L sgRNA into B-NDG mice. ORP4L deletion by Dox significantly reduced leukemia cell engraftment (Figure 6E) and improved the survival of the mice (Figure 6F). We then used the ORP4L-specific inhibitor LYZ-8120 to inhibit ORP4L. Overexpression of WT ORP4L but not ORP4L with LYZ-81 mutant binding sites (ORP4L M4)20 rescued primary ATL cells from death induced by LYZ-81 (supplemental Figure 5B-D), indicating the specific and direct cytotoxicity of LYZ-81 via targeting ORP4L in ATL cells. Moreover, treatment with LYZ-81 reduced AKT/NF-κB/p53 pathway (Figure 6G) and evoked death of the T cells of the patient with ATL (Figure 6H). To evaluate the tolerability of LYZ-81 in vivo, LYZ-81 (5.8 mg/kg every 2 days, IV) was injected into the B-NDG mice for 3 weeks. We assessed the mice’s weight every week and observed that LYZ-81 treatment had no effect on their body weight (supplemental Figure 5E). Histological analysis further revealed that LYZ-81 treatment did not alter the gross histology of spleen, liver, lung, and kidney (supplemental Figure 5F), implying that LYZ-81 is tolerable. We then tested the therapeutic effects of LYZ-81 in xenograft ATL models. LYZ-81 treatment (Figure 6I) further reduced the development of T-cell leukemia, promoted the survival of mice, and reduced the body weight loss induced by engraftment of T cells of patients with ATL (Figure 6J-K; supplemental Figure 5G). Finally, to confirm that xenografted cells were derived from ATL clones in our engraft ATL mode, we analyzed the HTLV-1 proviral genome of xenografted cells. The Tax region was amplified by PCR with total DNA derived from the spleen of xenografted B-NDG mice receiving T cells from 3 independent patients with ATL (supplemental Figure 5H), and with vehicle treatment, which generated xenografts containing human CADM1+ cell infiltration in the spleen (supplemental Figure 5I), demonstrating that HTLV-1–infected ATL cells become leukemic in B-NDG mice. These results strongly suggest that targeting ORP4L could be considered a promising approach for T-cell leukemia therapy.
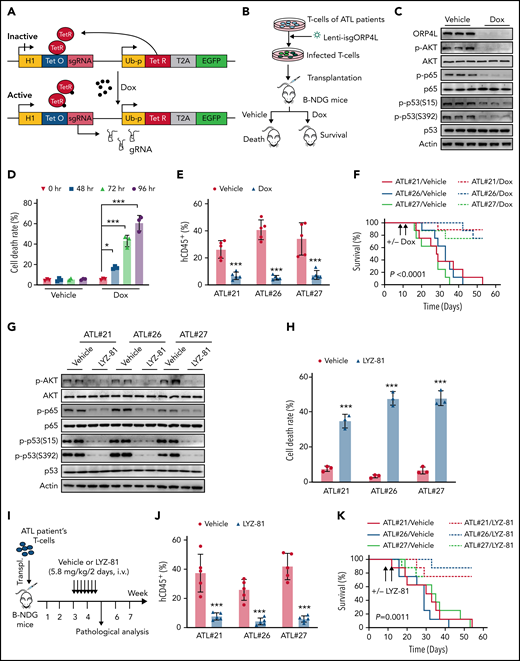
ORP4L inhibition results in T-leukemia cell death. (A) Schematic image depicting the Dox-inducible gRNA lentiviral vectors. Cas9 is constitutively expressed in the cells. Treatment with Dox rapidly induced the sgRNA expression, which activated Cas9 and directed it to the target genomic sequence. (B) The experimental design. T cells were obtained from patients with ATL and maintained in vitro during lentiviral transduction with vectors encoding constitutive Cas9 and the Dox-inducible ORP4L sgRNA. For the in vivo assay, cells were transplanted into B-NDG mice and then monitored for development of T-cell leukemia. To delete the ORP4L gene, Dox was administered via food pellets (625 mg/kg) 10 days after transplantation. (C) Western blot analysis of ORP4L knockout and downstream signaling pathway in the T cells of a patient with ATL (ATL#21) after Dox (1 mg/mL) treatment for 72 hours in vitro. (D) Cell death analysis of T cells in a patient with ATL (ATL#21) transduced with constitutively expressed Cas9 and Dox-inducible ORP4L sgRNA lentiviral vectors with or without treatment with Dox (1 mg/mL) for the indicated times. Mean ± standard deviation (SD; n = 3 experimental repeat; Student t test). (E) Percentage of human CD45+ T cells of patients with ATL in the peripheral blood of B-NDG mouse transplant recipients 10 days after treatment with Dox or no treatment. Mean ± SD (n = 5 mice of each group; Student t test). (F) Kaplan-Meier comparative survival analysis of B-NDG mouse recipients of human T cells from patients with ATL treated with Dox or not treated, as described in panel B (n = 8 mice each group, log-rank test). (G) WT/NF-κB/p53 signaling pathway in T cells of patients with ATL (ATL#21, #26, #27) after LYZ-81 (5 nM) treatment of 18 hours. (H) Cell death analysis of T cells of patients with ATL treated with LYZ-81 (5 nM) for 18 hours. Mean ± SD (n = 3 experimental repeat; Student t test). (I) The experimental design for LYZ-81 therapy after xenotransplantation of human ATL cells in B-NDG mice. Leukemia cells obtained from patients with ATL were transplanted into B-NDG mice. Three weeks later, the mice were randomly assigned to 2 groups and treated with the ORP4L inhibitor LYZ-81 (5.8 mg/kg IV every 2 days ) or vehicle control for 2 weeks. (J) The percentage of transplanted human CD45+ ATL cells in the peripheral blood of B-NDG xenotransplant-recipient mice after a 2-week treatment, with or without LYZ-81. Mean ± SD (n = 5 mice of each group; Student t test). (K) Kaplan-Meier comparative survival analysis of B-NDG mouse recipients of human ATL T-cell xenotransplants and treated with or without LYZ-81 (n = 8 mice each group, log-rank test). *P < .05; ***P < .001.
ORP4L inhibition results in T-leukemia cell death. (A) Schematic image depicting the Dox-inducible gRNA lentiviral vectors. Cas9 is constitutively expressed in the cells. Treatment with Dox rapidly induced the sgRNA expression, which activated Cas9 and directed it to the target genomic sequence. (B) The experimental design. T cells were obtained from patients with ATL and maintained in vitro during lentiviral transduction with vectors encoding constitutive Cas9 and the Dox-inducible ORP4L sgRNA. For the in vivo assay, cells were transplanted into B-NDG mice and then monitored for development of T-cell leukemia. To delete the ORP4L gene, Dox was administered via food pellets (625 mg/kg) 10 days after transplantation. (C) Western blot analysis of ORP4L knockout and downstream signaling pathway in the T cells of a patient with ATL (ATL#21) after Dox (1 mg/mL) treatment for 72 hours in vitro. (D) Cell death analysis of T cells in a patient with ATL (ATL#21) transduced with constitutively expressed Cas9 and Dox-inducible ORP4L sgRNA lentiviral vectors with or without treatment with Dox (1 mg/mL) for the indicated times. Mean ± standard deviation (SD; n = 3 experimental repeat; Student t test). (E) Percentage of human CD45+ T cells of patients with ATL in the peripheral blood of B-NDG mouse transplant recipients 10 days after treatment with Dox or no treatment. Mean ± SD (n = 5 mice of each group; Student t test). (F) Kaplan-Meier comparative survival analysis of B-NDG mouse recipients of human T cells from patients with ATL treated with Dox or not treated, as described in panel B (n = 8 mice each group, log-rank test). (G) WT/NF-κB/p53 signaling pathway in T cells of patients with ATL (ATL#21, #26, #27) after LYZ-81 (5 nM) treatment of 18 hours. (H) Cell death analysis of T cells of patients with ATL treated with LYZ-81 (5 nM) for 18 hours. Mean ± SD (n = 3 experimental repeat; Student t test). (I) The experimental design for LYZ-81 therapy after xenotransplantation of human ATL cells in B-NDG mice. Leukemia cells obtained from patients with ATL were transplanted into B-NDG mice. Three weeks later, the mice were randomly assigned to 2 groups and treated with the ORP4L inhibitor LYZ-81 (5.8 mg/kg IV every 2 days ) or vehicle control for 2 weeks. (J) The percentage of transplanted human CD45+ ATL cells in the peripheral blood of B-NDG xenotransplant-recipient mice after a 2-week treatment, with or without LYZ-81. Mean ± SD (n = 5 mice of each group; Student t test). (K) Kaplan-Meier comparative survival analysis of B-NDG mouse recipients of human ATL T-cell xenotransplants and treated with or without LYZ-81 (n = 8 mice each group, log-rank test). *P < .05; ***P < .001.
Discussion
In a set of mouse models, we showed that ORP4L is a prerequisite for T-cell leukemogenesis induced by HTLV-1 and is essential for survival of T-cell leukemia. We provide evidence of a novel initial pathogenic mechanism in which HTLV-1 induces T-cell leukemogenesis through a miR-31 loss/ORP4L gain axis. Enforced expressed ORP4L interacts with PI3Kδ to promote PI(3,4,5)P3 generation and subsequently activate PI3K/AKT signaling, resulting in oncogenic properties through NF-κB activation and p53 inhibition. Bioinformatics analysis of DEGs in LCK/R26Tax T cells, ORP4L-expressing T cells, and human ATL cells identified 3 DEGs, including CCNB1, CCNB2, and RRM2, that were involved in the p53 signaling pathway. Downstream targets of the p53 transcription factor mediate its different biological outcomes. p53 inactivation increases some pro-oncogenes, such as CCNB1, CCNB2, and RRM2, via inhibiting the suppressors of these genes 14-3-3 sigma and mTOR.34,36 Thus, p53 inactivation mediates the AKT/NF-κB signaling and upregulation of pro-oncogenes CCNB1, CCNB2, and RRM2 in Tax-induced T-cell leukemogenesis. Thus, we speculate that p53 mediates the AKT/NF-κB signaling and pro-oncogene misregulation in Tax-induced T-cell leukemogenesis.
Tax transcripts are detected in only ∼40% of all patients with ATL,4 yet most of these transformed ATL cells retain strong AKT activation, which suggests the existence of compensatory mechanisms when the Tax expression is silenced. Tax induces miR-31 loss in T cells with genetic and epigenetic abnormalities. Genetic loss at the miR-31 locus is observed in some cases of ATL. Moreover, the polycomb-dependent PRC2-mediated H3K27me3 was significantly and frequently reprogrammed, resulting in epigenetic regulation of multiple genes in ATL.29,37 Considering that genetic deletion is irreversible once it occurs, we speculate that this genetic and epigenetic regulation will lead to sustained oncogene misregulation and activation of the downstream oncogenic signaling pathway, even when the induction factor Tax is removed. Our results revealed that Tax-induced miR-31 genetic region deletion releases ORP4L expression that invokes AKT hyperactivation, NF-κB–dependent p53 inhibition and T-cell leukemogenesis. Thus, ORP4L is a compensatory factor that sustains continuous AKT/NF-κB/p53 oncogenic signaling in transformed T cells.
It is estimated that viral infections contribute to 15% to 20% of all human cancers.38 After acute infection, viruses reprogram cellular signaling pathways to establish persistent chronic infection that determines host cell fate. Meanwhile, host cell components, including lipids, play crucial roles in virus life cycles.39 Although studies have demonstrated that these virally modified cellular processes are governed by complex regulatory networks, the initial events of virus-induced cancer remain poorly understood. Given the role of the PM in signal transduction in response to environmental change, we speculate that remodeling of PM lipid composition may be the dominant contributor in carcinogenesis of virus-associated human cancers. Moreover, host lipids and their synthesis may affect viral infectious process.40 For instance, OSBP is crucial for the HCV maturation process.41 Further studies are needed to investigate in depth whether the ORP4L machinery also contributes to the various steps of the HTLV-1 virus life cycle.
We showed that engineering ORP4L expression alone is sufficient to initiate the development of T-cell leukemia in a mouse model with clinical and pathological features similar to human ATL, uncovering a novel molecular event associated with HTLV-1–induced T-cell transformation. New anticancer drug development efforts aimed at identifying metabolic codependencies must consider not only targeting and inhibiting tumor cells, but also avoiding off-target impact on normal cells.42 ORP4L expression is restricted in normal tissues,43 but is activated and plays an essential role in survival of T-cell leukemia, raising the possibility of a promising therapeutic window for the development of ORP4L inhibitors. Acute ATL is extremely resistant to conventional chemo/radiation therapy. Our data demonstrate that LYZ-81 eliminates ATL cells in vitro and in vivo via inhibiting ORP4L, suggesting this compound as a starting point for drug development for ATL therapy.
Acknowledgments
This work was supported by National Science Foundation of China (NSFC) grant 32071280 and 8177043 (D.Y.), Major Research Program of Guangdong Science and Technology grant 2017A030308002 (D.Y.), and NSFC Grant for Young Scientists of China 31900548 (W.Z.).
Authorship
Contribution: D.Y. conceived and designed the study; data were collected and analyzed by W.Z., X.C., G.P., Q.N., X.F., M.X., and M.L.; W.Z. and D.Y. wrote the manuscript with the assistance of Y.H. and Q.Y.; and all authors commented on the manuscript, data, and conclusions.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Daoguang Yan, Jinan University, 601 Huangpu Ave, Guangzhou 510632, China; e-mail tydg@jnu.edu.cn.
Original data may be obtained by e-mail request to the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.